Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by debbie
| Link to this post | posted 03 Apr, 2019 02:23 | |
|---|---|

|
Hi! Wow! More details are needed. Would you provide who you are, Where you are from, What host you are using? Once I get some basics, I may have more questions. |
| Link to this post | posted 28 Mar, 2019 23:51 | |
|---|---|
|
|
Joann, I do't believe a sequence has to be very long to contain a transmembrane domain. for me the 2 sides of the coin are: 1. is one membrane real? (so we ask that you can find it with 2 different finder programs) 2. It appears that defense systems (toxins, for example) can be 'attached' to the cell membrane, so that membrane protein might be a good place to look into that if you have a phenotype that suggests that phage has provided a defense. i.e. it would just be good to know. It will also be found out once you have a phenotype to investigate, so there is no harm by not calling it. Not helpful, right? So, I think my take on this is if you take the time look and confirm, report it. If this is all you miss in the genome, it is small potatoes. |
Posted in: Functional Annotation → Membrane protein
| Link to this post | posted 26 Mar, 2019 19:01 | |
|---|---|
|
|
Cool! |
Posted in: Cluster A Annotation Tips → Pham 23651 function assignment
| Link to this post | posted 26 Mar, 2019 19:00 | |
|---|---|
|
|
Joe, I think you are confusing apples and oranges here. The 'standard code" settings have no effect anywhere in auto-annotation. Also if you have chosen ATG, GTG, and TTG start codons in the Translation tab of the Local Settings in your preferences, DNA Master will display them. You can select "Bacteria and Plant Plastid Code" in the New Features Tab of the Local Settings. Is that what changes? What is changing on you? debbie |
Posted in: DNA Master → DNAM failing to use proper translation code
| Link to this post | posted 26 Mar, 2019 18:52 | |
|---|---|
|
|
I still like my little gene call. This area is definitely tricky. |
Posted in: Gene or not a Gene → Forward or reverse gene?
| Link to this post | posted 26 Mar, 2019 08:40 | |
|---|---|
|
|
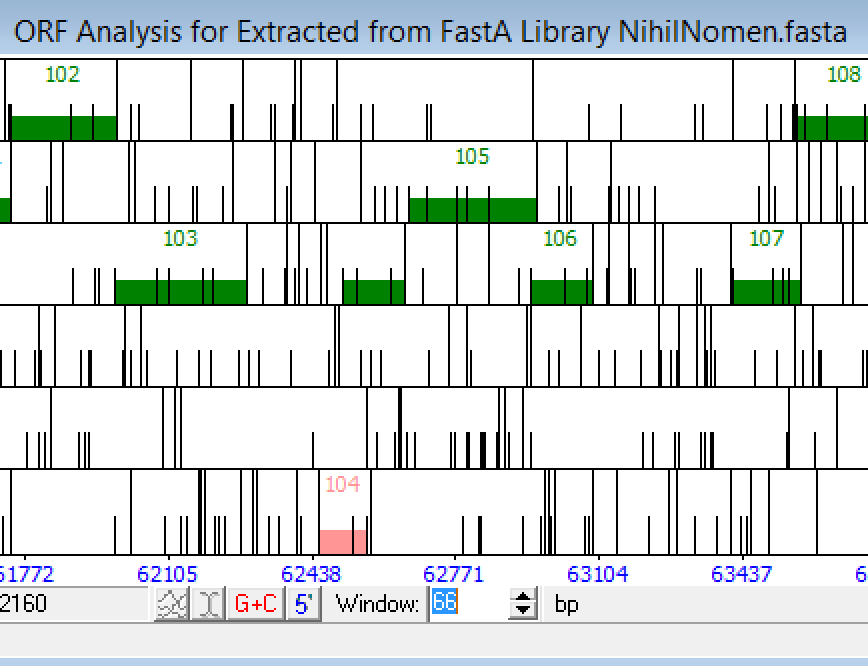
Philippos, Hi. No, I would not call auto-annotated gene 123, but instead call the longer gene you describe (it is a DNA methylase, so that should convince you). In addition, don't miss the next forward gene, because it too is a DNA methylase. See my attached frames window. Check out the differences in the auto-annotation…. |
 25Kb
25Kb| Link to this post | posted 26 Mar, 2019 08:27 | |
|---|---|
|
|
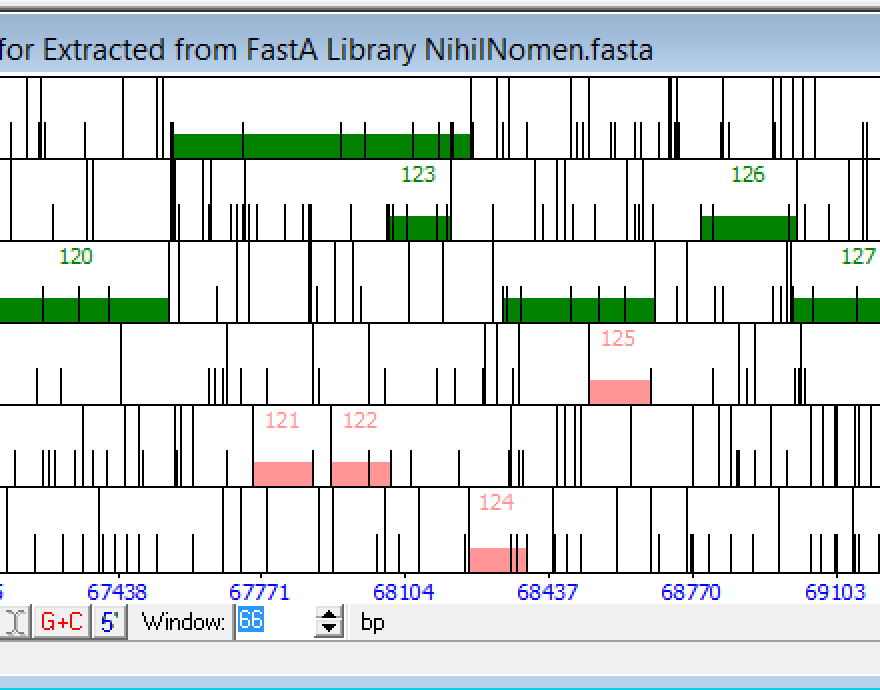
Philippos, Hi. I took a look at this area and I would not call that or the next reverse gene(see Phamerator). Instead I wold add a small ORF in the forward direction between 103 and 105. If you look at your auto-annotation, my auto-annotation, and Phamerator, I think there are differences in this region across all 3. Which means basically there is poor coding potential all-around. Note: I see no strong coding potential in the reverse frames. The red dotted lines on a GeneMark graph are demonstrating an order of 2 (patterns of 2 nucleotides at a time), which most of the time amounts to noise. So if you look at your example, the black line is poor coding potential (order of 4). Make sense? debbie |
 26Kb
26KbPosted in: Gene or not a Gene → Forward or reverse gene?
| Link to this post | posted 21 Mar, 2019 11:53 | |
|---|---|
|
|
Sarah, After preliminary work with host range experiments, I am finding that the best way forward is to find phages on hosts that you would like to test. Until you are familiar with a host, the testing of additional hosts with known phages can be problematic. By that I mean, if you don't have a phage that infects each host, how do you know that a phage that you are testing can infect that host (or that that infection is detectable)? If it does, great! But if it doesn't, is your test valid (without a positive control)? So my recommendation going forward is to add some new hosts, go phagehunting on that host, then begin the host range testing. Does that make sense? |
Posted in: Host-Range Project → Basic Host Range Project Information
| Link to this post | posted 18 Mar, 2019 18:45 | |
|---|---|
|
|
Veronique, I agree, I see no function to call for the second gene in question. and I would call the first one the "capsid maturation protease and MuF-like fusion protein". |
Posted in: Functional Annotation → capsid maturation protease
| Link to this post | posted 15 Mar, 2019 02:54 | |
|---|---|
|
|
Erin, I don't think it has to have 2 domains, it just has to have 1 convincing one. The problem is that one program doesn't have adequate sensitivity. So if there is only one, we want it verified by 2 sources. Without the option to 'verify' a transmembrane domain with a second program, let's not add it as a function. |
Posted in: Functional Annotation → Membrane protein