Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by DanRussell
| Link to this post | posted 21 Apr, 2016 19:23 | |
|---|---|

|
Aaron Stevens Hi Aaron, Just sent them. Let me know if they don't come through or don't work! –Dan |
Posted in: SEA-PHAGES Virtual Machine → Student download of 2016 VM
| Link to this post | posted 13 Apr, 2016 19:08 | |
|---|---|
|
|
Another slightly more complicated way would be to upload it to something like Google Drive or Dropbox from within the VM (using Chrome), and then download it outside the VM. –Dan |
Posted in: Starterator → Saving Starterator reports outside of VM
| Link to this post | posted 13 Apr, 2016 18:08 | |
|---|---|
|
|
cmageeney Hi Katie, Just emailed them to you! Good luck. –Dan |
Posted in: SEA-PHAGES Virtual Machine → Student download of 2016 VM
| Link to this post | posted 11 Apr, 2016 14:04 | |
|---|---|
|
|
Steven Felkoski Yep, Steven, just emailed them to you. –Dan |
Posted in: SEA-PHAGES Virtual Machine → Student download of 2016 VM
| Link to this post | posted 06 Apr, 2016 20:16 | |
|---|---|
|
|
Hi Lee, Looks like that error is my fault! When I updated the consed on the VM, apparently I didn't copy all files from the old consed directory to the appropriate places. This would only affect uses of phred and phrap, which are rarely used, but would definitely be used when adding reads. To fix this on your VM, run the following commands when logged in as seafaculty, entering the password as necessary: This should put the files in the appropriate places and make it work again. You're the first to discover this probably because 1) we barely do any Sanger reads any more and 2) I usually run consed on my Mac rather than within the VM. Sorry, and good catch, –Dan |
Posted in: Consed → Adding Sanger Reads
| Link to this post | posted 04 Apr, 2016 18:41 | |
|---|---|
|
|
Hi Greg, Looks like in that paper we mention using ClustalW which has been retired in favor of ClustalOmega to generate the numbers, then NJPlot to draw the trees. –Dan |
Posted in: Papers → Pham and Phylogenetic analysis software
| Link to this post | posted 23 Mar, 2016 18:16 | |
|---|---|
|
|
ball.1766 Hey Sarah, If he's just installing Phamerator on his own Linux system, then he probably just needs this quick guide rather than any pre-made image/file or anything. It's not too tough to get it up and running on Linux. –Dan |
Posted in: SEA-PHAGES Virtual Machine → Student download of 2016 VM
| Link to this post | posted 07 Mar, 2016 15:25 | |
|---|---|
|
|
Hi Greg, If you really want a quick way to check your phages against some others, you could set up a new Phamerator database using the public version of PhamDB we've set up. There's some more info here. I should mention that I would keep all PhamDB databases relatively small for now, fewer than 100 genomes for sure. –Dan |
| Link to this post | posted 07 Mar, 2016 15:23 | |
|---|---|
|
|
Hi all, Ever wanted to just make your own Phamerator database, but then looked at all the necessary code and immediately given up? It's not an easy process, for sure, but a student named James Lamine from Calvin College—under the supervision of Randy DeJong—has made it much easier. Some of you may remember Randy's talk at last year's Symposium about PhamDB, a web-based way to make your own Phamerator databases. It takes GenBank (.gb) files as input, and outputs links that you can point your copy of Phamerator to in order to use the new database you've created. PhamDB has now been published in Bioinformatics. The installation instructions to get it set up and running on your machine are not bad at all. But we've also set up a (sort of) public version of PhamDB if you want to give it a try. Because it doesn't have account-specific permissions yet, we didn't want to make it fully public, lest random people delete your new databases. So if you're interested in using it, just email me (dar78@pitt.edu) and I'll send you the relevant info. Here's a screenshot: 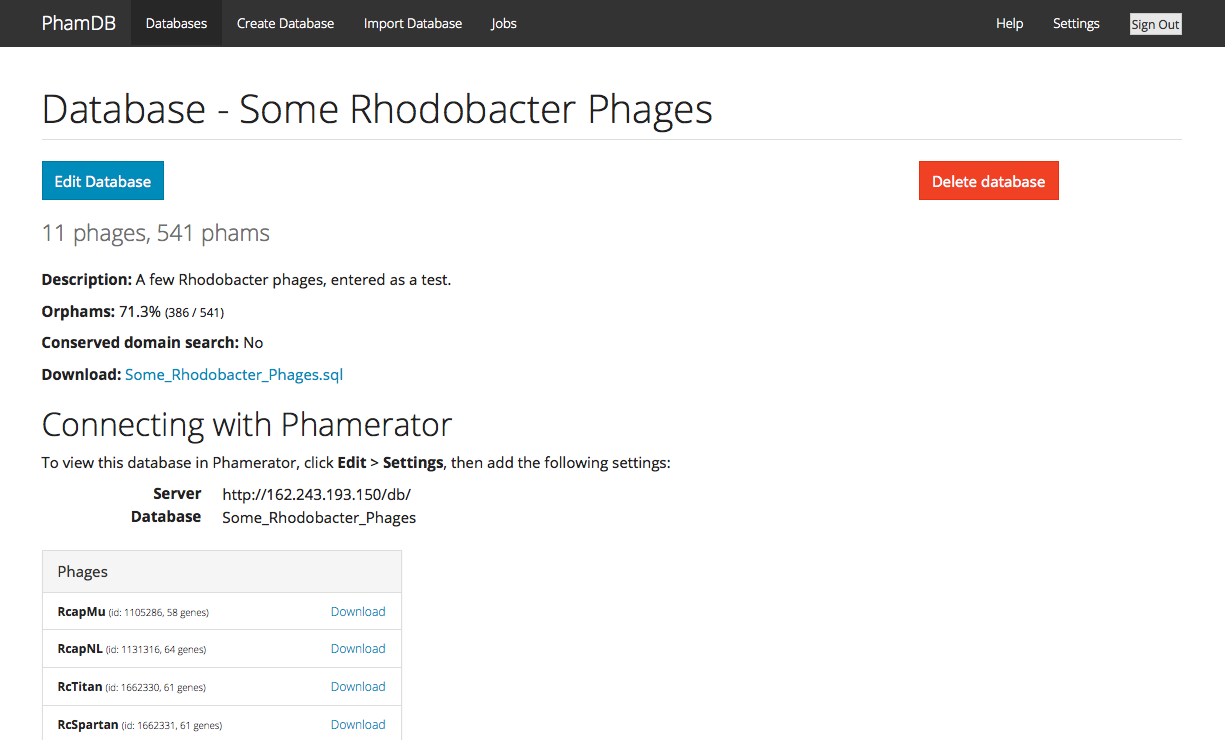 –Dan |
Posted in: Phamerator → PhamDB: Make your own Phamerator databases
| Link to this post | posted 07 Mar, 2016 15:09 | |
|---|---|
|
|
Hi Stephanie, Sure! First let me say that it’s always possible that any two auto-annotations on the same exact sequence won’t match 100%. This is because of the way the algorithms work and databases change, so if you had 25 students each run an auto-annotation they might not all match. But…the more likely explanation in your case is that it appears that Watson potentially has 3 tRNAs right after gene 8. Since Phamerator doesn’t show tRNAs, you just see a small gap there in Phamerator, though the Phamerator numbering does include those, so it looks like it skips from gene 8 to gene 12. DNA Master doesn’t number the tRNAs by default, so the first gene after them ends up being #9 (which was #12 in Phamerator). You can kind of see the Phamerator and DNA Master maps lined up below. 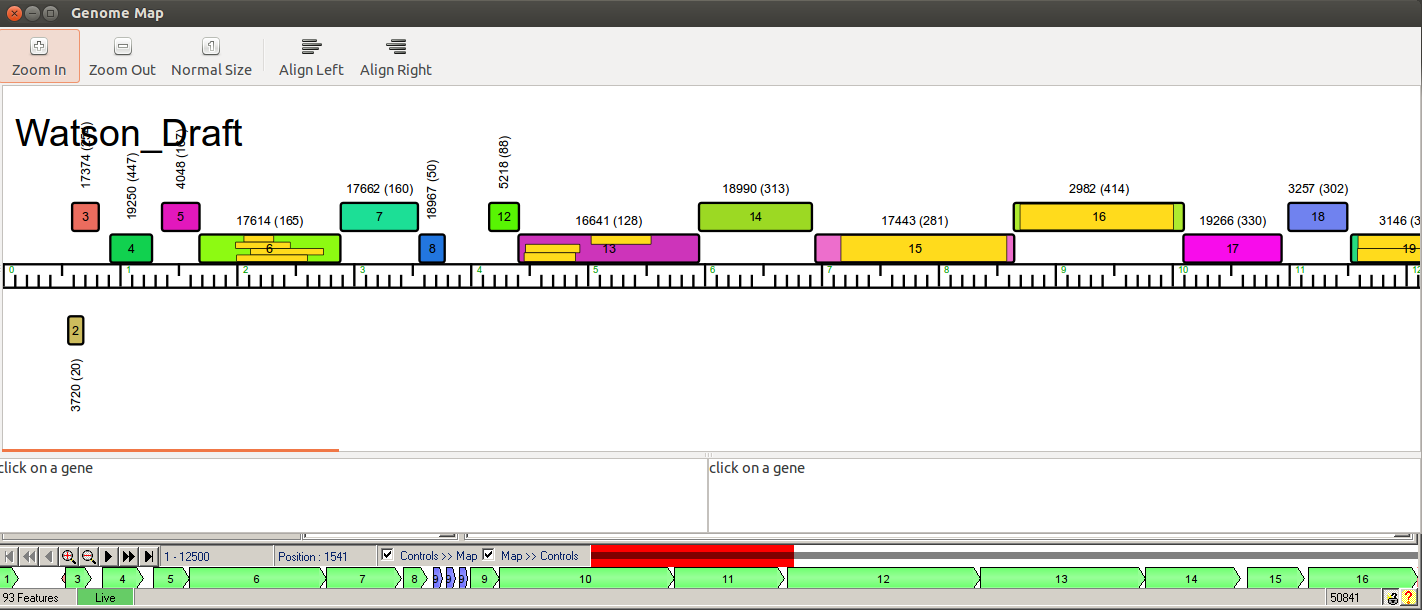 Because of this, we never really work by gene number. We instead use start/stop coordinates to identify genes, because those are unambiguous. And if you add or delete a gene, the gene numbers will eventually change anyway to accommodate the new/deleted genes, so they’re ephemeral until the very final submission. Hope that helps! —Dan |