Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by cdshaffer
| Link to this post | posted 05 Feb, 2020 20:13 | |
|---|---|

|
I am adding an online link so it is much easier to see the current Starterator database version. This link should always give the most recent database version that has been published: http://phages.wustl.edu/starterator/database.version |
Posted in: Starterator → Database Version for starterator
| Link to this post | posted 05 Feb, 2020 16:20 | |
|---|---|
|
|
Yes a typo of any kind can keep the system from changing to a green check. One simple error that is hard to detect is if there is a space character to the beginning or end of the function. The other thing worth mentioning but cannot explain your high rate is that the approved function list changes quite quickly but Pecaan only updates its list periodically. So again you can have synchronization issues. It is not uncommon for us to have an X or two by the end of an annotation because of this. Not sure why you are getting up to 1/3rd. So my suggestion is the same as JoAnn, encourage the students to type just a few characters in the function and then select the correct entry from the dropdown. |
Posted in: PECAAN → PECAAN Down?
| Link to this post | posted 03 Feb, 2020 03:41 | |
|---|---|
|
|
This is all happening because of database synchronization issues. Right now pecaan is on 336 and the rest of us have already updated to version 337. You can tell which version in various ways. Since phagesdb.org is always in sync with the web server that publishes the database, you can bookmark this page to see the most current version of the database: http://phamerator.webfactional.com/databases_Hatfull/Actinobacteriophage.version For phamerator.org there is a pulldown menu in the top right with the database version number; for Pecaan the version is noted just above the map on the Pham Maps tab; for starterator the version is noted just after the run date near the top of the text section of the report so any working report can be used. You can also just go to the URL "phages.wustl.edu/<version>/" to see if that version has been posted. For example, to check if version 337 is available go here: http://phages.wustl.edu/337/ All those reports are specific for version 337. Since Pecaan is on an older version than starterator there is a work around. Just change the "starterator" in the url to the database version number. So when pecaan is using the old 336 and it tries to use an out of date link, this one for example http://phages.wustl.edu/starterator/Pham99984Report.pdf it fails with a 404, so knowing Pecaan is using version 336 replace the "starterator" in the URL with the version number like this: http://phages.wustl.edu/336/Pham99984Report.pdf.pdf and now it works. When starterator is behind pecaan there is not much that can be done but wait until I post the newest set; but please feel free to send me an email and I can at least give you an ETA. P.S. the above links may not work correctly once version 338 of the database is released. |
Posted in: PECAAN → PECAAN Down?
| Link to this post | posted 29 Jan, 2020 22:27 | |
|---|---|
|
|
To adjust the VM desktop image use the virtual box View -> Virtual Screen -> Scale to… menu item. When I am on a high res screen like my macbook pro screen I set "Scale to 200%" and when I am on an external monitor I set "Scale to 100%" but feel free to pick whatever scale works best for you. |
Posted in: Using WINE to run DNA Master on a Mac → Help with WINE
| Link to this post | posted 21 Jan, 2020 23:15 | |
|---|---|
|
|
The picture you posted looks like you are running an old version of DNA Master. Mine version of that tab looks like the attached. You should double check that you have an up to date version of DNA Master. I just checked and it says: Version 5.23.3 build 2693 from 26 November 2019 which it reports is up to date. |
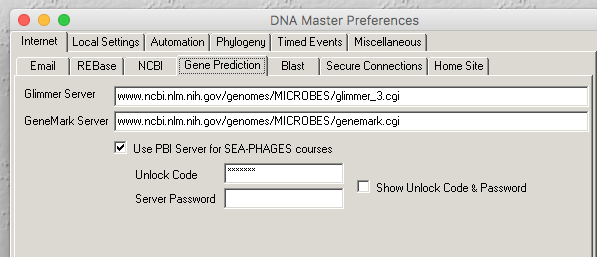 27Kb
27KbPosted in: DNA Master → Auto-annotation fix for fall 2017 and later
| Link to this post | posted 21 Jan, 2020 23:06 | |
|---|---|
|
|
With the MLK holiday weekend I was not able to calculate the new reports until this afternoon for the recently posted version of the database. The reports for this release of the database (number 336) has now been posted and pham 106383 is now available. Most missing phams are due to this issue of databases being out of sync. Unfortunately, it takes up to 20 CPU hours to calculate all 14,000 or so Starterator reports. Thus Starterator will typically be the last to update and often up to 1 day later than the other web pages. You can see which version of the database PECAAN is using on the PhamMaps page just above the map; for phamerator.org you can see which version in the pull-down menu near the top left corner; for Starterator the database version is now indicated near the top of the second page just after the date run. If these numbers do not match you can expect a higher risk of missing phams until the new Starterator analysis has been posted. |
Posted in: Starterator → Pham 106383 report not found
| Link to this post | posted 18 Dec, 2019 00:12 | |
|---|---|
|
|
Sorry about that. Yes, you unfortunately got stuck in the awkward time between when the new version of the database gets uploaded and all the Starterator reports get published to the web. Starterator has the most processing to do on each pham so it will always be the last piece to update. I was able to push the most recent reports to the server late this morning after an overnight run. As the database grows it is taking longer and longer to process all those pham reports and it is now often taking up to 1 full day to get things uploaded. |
Posted in: Starterator → Pham not showing up
| Link to this post | posted 09 Dec, 2019 19:35 | |
|---|---|
|
|
it does not appear that the large overlap is found in the BD2 cluster. Large overlap is found in most other subclusters. |
Posted in: Cluster BD Annotation Tips → DNA Primase
| Link to this post | posted 15 Nov, 2019 21:24 | |
|---|---|
|
|
This is to announce the release of Starterator version 1.2 This release has some graphical updates and incorporates cluster information into the text reporting. The cluster information can be particularly helpful for large complex phams but has little or no value for small phams comprised of a single cluster. The new version will be used to create all online pham reports starting with database version 233 which should be posted to the web based pham reports the afternoon of Nov 15th. For anyone still using the virtual machine the new version will be made available soon for download and update (details to be posted soon). The main new features in this update: 1. Tracks now display only that region of the genes with starts, and, if the tracks are judged to be highly complex, the image is further zoomed in to show only the region surrounding called start sites 2. Tracks are now grouped by subcluster and the color of the track changes with each subcluster (Thanks for the idea go to Sally Molloy) 3. Cluster information of each phage is now added to the "Summary by Start" section (Thanks for the feature request go to Claire Rinehart) 4. A new "Summary by cluster" section has been added to give info on manual annotations specifically for each cluster found in the pham 5. Details on the number of manual annotations (MA's) for each start in a gene are now given in the "Gene information" section. Also added to the whole phage report (available if you still run starterator in a VM): 6. A new summary table on the analysis of manual annotations of pham members and the degree of consensus with the annotated start. 7. Simplified reporting focusing on the relevant results for the phage in the whole phage report 8. Support for parsing of the "CDS function" file exported from Pecaan when using the whole unphamerated phage analysis |
Posted in: Starterator → Release of Starterator version 1.2
| Link to this post | posted 03 Oct, 2019 16:10 | |
|---|---|
|
|
down for me as well when since last Monday |
Posted in: tRNAs → Aragorn Issue