Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by cdshaffer
| Link to this post | posted 03 Oct, 2017 20:53 | |
|---|---|

|
I can confirm your results, when I run yeezus through the pecaan auto-annotation I get 86 gene prediction. Looking at the gene list on phagesdb I see 93 gene calls. Looking at it I can see for example that yeezus gene 7 called in phagesdb is missing from the pecaan auto-annotation. This difference in number of gene calls is likely due to minor differences in the settings used when running glimmer and genemark at the two different sites. When I run glimmer and genemark on my local machine I often get different results than what DNA Master got from the NCBI glimmer and genemark servers. Since the starterator results are based on the gene calls done by phagesdb, gene numbers will quite likely get out of sync with your DNAMaster file. Your best bet is to switch over to the new auto-annotation server inside DNA Master. There was an email with the instructions a couple of weeks back but here is a link to the pdf with instructions on how to get auto-annotation working in DNA Master. I believe once you set it up, you can just re-run auto-annotation in DNA Master and you should get the same 93 genes that are in phagesdb and starterator. |
Posted in: DNA Master → Glimmer Failure on Auto Annotation
| Link to this post | posted 27 Sep, 2017 18:44 | |
|---|---|
|
|
You can't really "merge" the two databases since mysql will just use pham numbers to decide whether to put proteins in the same pham. You want all similar proteins from both phage collections to be put in the same pham when they are similar to each other. The only way to do that is to rerun all the protein comparisons with all the proteins in the database. There is a thread on how to make databases using a web based program called phamDB. See this thread. The program is old enough now that the installation instructions are likely out of date and it will take some tweaking to install but the basic code is still sound. Alternatively, you could use K_phamerate scripts to create your database, you need to check out and use the code at the sea-phages github repository. That repository holds the code currently being used to create the phamerator databases and is being actively maintained by Travis Mavrich. Unfortunately you will have to install and run using command line. |
Posted in: Phamerator → Phams
| Link to this post | posted 22 Sep, 2017 16:27 | |
|---|---|
|
|
Too bad none of the easy work arounds helped. I am going to assume that there are no important files inside the SEA VM, do NOT do the following if there are important files you need to recover from inside the VM. If you have files to recover you are going to need local expert who can work with your computer to get Ubuntu to boot so the files can be recovered, could be a long and arduous trial and error process of fixing Ubuntu. So assuming there are no files to recover, I would try to fix it by simply deleting and replacing the SEA VM. Sorry I don't use Virtualbox in Windows so some of this might be a little off but should suffice: 1. Open virtualBox program so you get something like this window (i think the pic is from mac, the windows version should look similar): 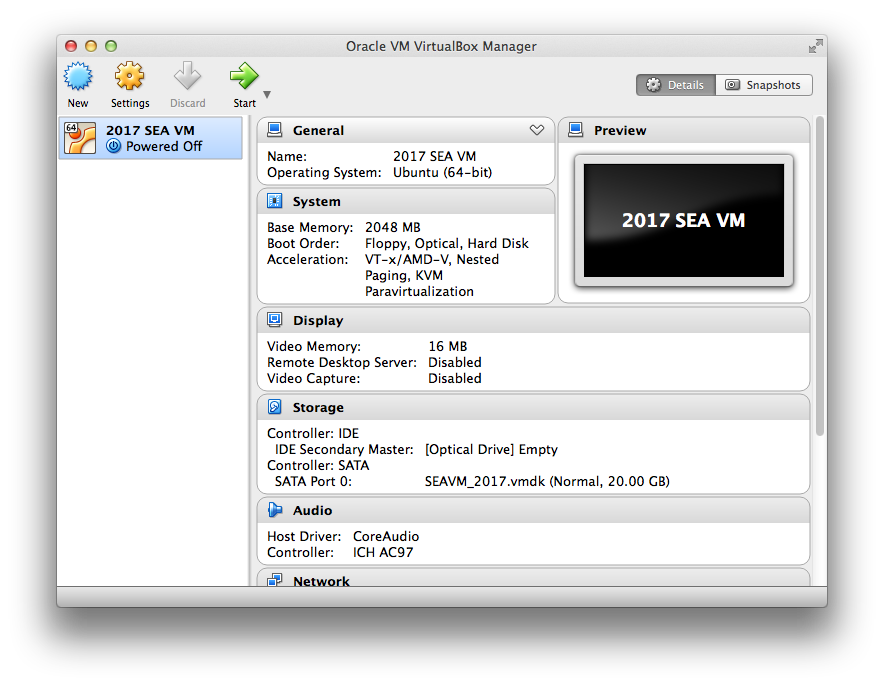 2. click on the "2017 SEA VM" in the left column and select "Remove…" from the machine menu 3. You should see a message about how you are "about to remove a machine from the machine list" and three buttons. Select "Delete all files" to completely remove and delete the SEA VM and all associated files 4. Go back to https://seaphages.org/software/virtualmachine/ and start with step two to reinstall the SEA VM, booting into Ubuntu and installing the guest additions. |
| Link to this post | posted 21 Sep, 2017 16:14 | |
|---|---|
|
|
Few things to try and report back: 1. Can you ignore the box, and just start phamerator? 2. What happens if you just click the red close button at the very top left? Does it still kick you back to the login screen? 3. Does this happen on both the student and faculty accounts? |
| Link to this post | posted 22 Aug, 2017 16:29 | |
|---|---|
|
|
Sorry about that, we are having network upgrades here that have made it difficult to run the starterator updates. Until these are fixed starterator may lag a bit more than usual on when updates get posted. There was also a rollback of the phamerator database from version 141 back to version 140. I just rolled the starterator reports back so all the reports at phages.wustl.edu should now be in sync with version 140 which was run on 8/14/17 and should match phagesdb and phamerator.org. |
Posted in: Starterator → Updates to Starterator output
| Link to this post | posted 18 Aug, 2017 22:14 | |
|---|---|
|
|
just to document for anyone what wants to create an auto-annotation here is what I did: 1. open DNA Master and do the standard File -> Open Fasta and import the sequence 2. Go go https://discover.kbrinsgd.org/autoannotate/ and enter phage name and upload the same sequence file you used in step 1 3. Click "Process" 4. In the text box below the process button you will get the auto-annotation as "Documentation"; it will have a bunch of entries that look something like this: 5. Select and Copy the entire contents of that text box 6. Go back to DNA master, on the phage window click the "Documentation" tab at the top right 7. Delete any text in the large text box and paste in from the results from the PECAAN auto-annotation. (When I did this this step I got about 100 question marks at the end which I deleted so the last entry ended like this: 8. click the parse button, then in the window that opens accept the defaults and click the new parse button. This will convert the "documentation" into DNA Master gene entries 9. Save the DNA Master file as usual |
 /gene="232"
/product="gp232"
/locus tag="Roots515_232"
/note=Original Glimmer call @bp 155929 has strength 11.01 ** not called by GeneMark
/gene="232"
/product="gp232"
/locus tag="Roots515_232"
/note=Original Glimmer call @bp 155929 has strength 11.01 ** not called by GeneMark
Posted in: DNA Master → Glimmer Failure on Auto Annotation
| Link to this post | posted 17 Aug, 2017 16:00 | |
|---|---|
|
|
I just tried to run the update on my Windows 7 machine and it worked fine, so yes the ftp server appears to be up so that is not the issue. Sorry but not a Win 10 expert but I think you are right is is some kind of setting on the pro version, likely for heightened security |
Posted in: DNA Master → FTP Error on Windows 10 Pro
| Link to this post | posted 14 Aug, 2017 17:47 | |
|---|---|
|
|
Likely a database update issue since I was not able to login and update the database for about a week. I just posted the most recent version of the database to the starterator website. Check now and post again if still missing. |
Posted in: Starterator → Updates to Starterator output
| Link to this post | posted 19 Jul, 2017 17:12 | |
|---|---|
|
|
It is not you or your system. I am getting the same result. It looks like NCBI is no longer serving glimmer and genemark analysis as a web page (which is what DNA Master used) and is now forwarding all web requests for those pages to the download pages of the software. This totally breaks DNA Master auto-annotation as you have discovered. A long term solution is going to require updates to DNA Master. In the short term, it is probably easiest to use PECAAN. You can start here: https://seaphages.org/forums/topic/224/ Which explains how to get an account and a link to the user guide. PECAAN is under very active development so the user guide will help get you started but expect the actual web pages to be different. Pecaan is nice in that it will do a lot of the drudgery of entering notes in a proper format if you use it to do all your annotations. So you may want to consider switching over to using it for a lot of the annotation work. If you are in a huge hurry (like you need the DNA Master file ASAP for a class) just post the name of the phage I can use pecaan to generate the auto-annotation file for you very easily. If you want to try to use pecaan to do most of your annotation, feel free to post any follow=up questions to the PECAAN section of the forum. |
Posted in: DNA Master → Glimmer Failure on Auto Annotation
| Link to this post | posted 03 Jul, 2017 16:55 | |
|---|---|
|
|
I would go with the sequence as is then (i.e. 6 G's). The sequencing error hypothesis was always a bit of a long shot since the consensus was too long and as I mentioned 454 typically makes consensus errors by making mononucleotide runs too short. As for annotation, that is up to Veronique & Welkin. |
Posted in: Gene or not a Gene → Gene split in 2