Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by cdshaffer
| Link to this post | posted 07 Apr, 2023 17:35 | |
|---|---|

|
TL;DR: I see on Pecaan that your current annotation is "DNA Primase/helicase" and I would also annotate that way as well. Long story: I ran a full HHPRED result and looked at the overall general structure of this protein. See attached annotated imare. I noted in the results 3 regions each with a unique set of hits. In looking at the N terminal region the consensus of the 4 hits in the green box strongly suggest some kind of primase or polymerase. Since primases are a specialized type of polymerases cannot really tell from just those 4 hits what would be the best annotation. However the middle section has over 100 hits and I cut off the image with the top 15 or so. The general consensus of all those hits in the blue box are (as you said) some kind of helicase which is also supported by the AAA-ATPase hits. The really curious part is all the hits in the region of the orange box, the general consensus of all those hits is a DNA binding domain. I don't ever remember seeing a DNA binding domain attached to a helicase domain. I did look through the pfam architectures to see if this combination has been seen before but my 10 minutes of poking did not reveal any examples like this. So taken together we have a DNA binding domain, a helicase domain, and a primase/polymerase domain. It looks to me like maybe a polymerase specifically designed to start replication at a single location "maybe?". It would take a really deep dive to know for sure this combination of domains is truly novel but with just my 30 minutes poking around I could not find anything quite like this, hence I have no good annotation because there is nothing else like this I could find in the published literature to link to. So the practical solution in my mind is to pick the best of the approved terms, which in this case is to highlight the presence of both the primase domain and the helicase domain. Hence I would annotate as you did: DNA primase/helicase As with many annotations, not ideal, one might say "not even good" but in the end it is the best term we have. |
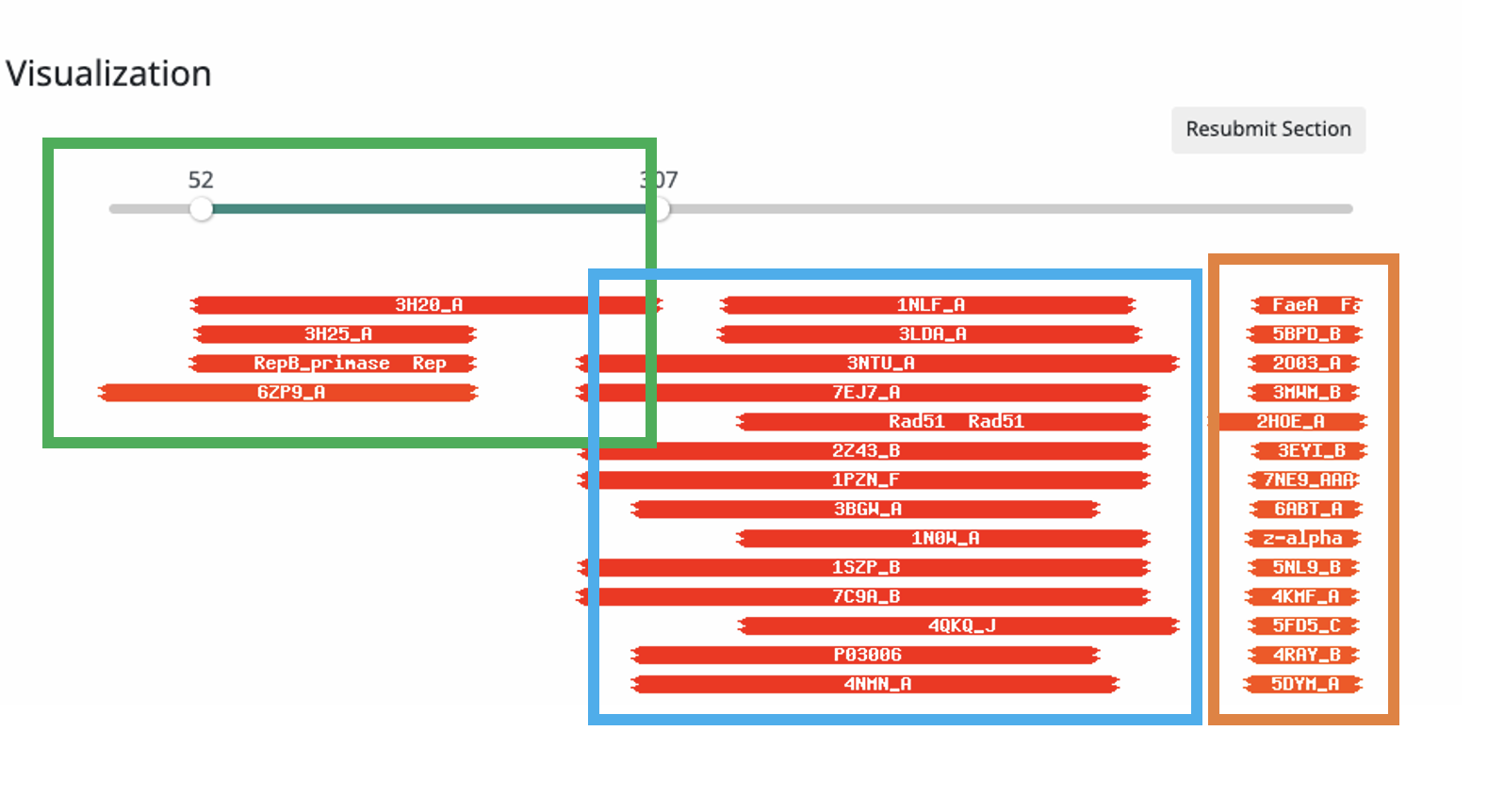 206Kb
206KbPosted in: Functional Annotation → GG cluster DNA primse/helicase
| Link to this post | posted 16 Mar, 2023 21:08 | |
|---|---|
|
|
I would discount the observation that "majority of the functions as hypothetical" because most of those "hypothetical proteins" were probably annotated before the above discussion and thus before the addition of "helicase loader" to the approved terms. So in this case, "absence of evidence is NOT evidence of absence". New crystals are constantly being published and the approved terms list is a living document that changes as we find new functions and fine tune the nomenclature. This is one of those places where I would tell a student "This is why we keep doing annotation by hand, we keep learning more and more and we keep getting better and better at annotation". Looking at the positive evidence though, this call is indeed tricky, there are several HHPRED hits suggestive of helicase loader that all have really high probability but only about 40-50% coverage. So this is where reasonable annotators can disagree. In looking at the crystal data here I can see that the part that does not align is "disordered" so one could use that to argue that a strictly similar structure in this region is not required for function (as this region is not highly structured in the crystalized protein) and thus the fact that it does not match at the structural level is not good evidence that this new protein is not another example of a helicase loader. Bottom line, the fact that this region is disordered means that I discount the evidence that HHPRED is not matching them (i.e. it weakens the negative result). I don't think I ever like adding annotations on just a single piece of weak evidence, even if I can make a handwavy argument for why it is weak, so I would want more evidence. My own sense would be to look for synteny evidence to strengthen the call for a helicase loader. Since proteins that interact are much more often found near each other in phage genomes, you might find some positive results that give you more confidence you have a helicase loader. Is this gene near other genes that look to be part of a replisome or near some type of helicase? If you find a nearby helicase then you have found additional evidence. Synteny is never strong evidence, but combining two pieces of weak evidence (synteny and partial HHPRED), can sometimes clearly provide sufficient evidence and give you confidence to "make the call". |
| Link to this post | posted 03 Mar, 2023 16:45 | |
|---|---|
|
|
Unfortunately, being an "Orpham" means that the protein has not other similar proteins and is thus placed in its own pham group. Starterator is about using evidence from evolution to help gather evidence for start codons based on conservation and evolution of the genes by comparing how they have evolved over time. However for orphams, with only one member in the pham, there is nothing to "compare", so there is nothing to report. Start codon choice will just have to proceed without evidence from comparative evolution and rely on what evidence is available. |
Posted in: Starterator → Pham not found in Starterator
| Link to this post | posted 02 Mar, 2023 17:11 | |
|---|---|
|
|
OK DeepTMMHMM is working for me again but I had to create an account. It was still failing when I tried to use DeepTMMHMM as a guest. I used my github account to sign in thru OAuth but it looks like you might be able to just create an account de novo with an email address. |
Posted in: Functional Annotation → Deep TMHMM?
| Link to this post | posted 01 Mar, 2023 21:55 | |
|---|---|
|
|
Not working for me on web page either. I did not test command line, but the error log for my web run shows that the program was not able to initialize at the biolib cloud service. Unfortunately, the web page and the command line both submit to biolib so if biolib is down not much can be done until they fix the service. I cannot seem to find any way to get the status of the biolib cloud service so if anyone knows how to that, would love to hear it. There is a way to run the whole analysis locally but it requires access to specialized GPU hardware to do the analysis so we are really just stuck waiting for biolib service to be restored. Hopefully they are aware of this issue and are working on a solution, again can't find a status page anywhere, so hard to know. |
Posted in: Functional Annotation → Deep TMHMM?
| Link to this post | posted 29 Jan, 2023 22:44 | |
|---|---|
|
|
Just an FYI, I have been trying to reach PECAAN all day today with no success. Also, this website is also reporting it is down. Emails and messages have been sent to Clair and Dex. |
Posted in: PECAAN → PECAAN Down?
| Link to this post | posted 11 Jan, 2023 20:24 | |
|---|---|
|
|
when presented with this issue of specificity (i.e. should one annotate using the more specific term that includes the "MerR like" ) I like to first check out the PFam hits; others prefer the CD hit database, both are fine, I am just used to the Pfam dataset and I think the Hidden markov models that underlie PFam database (instead of the PSSM data in CDD) are more sensitive. You can add the Pfam database to an hhpred search, I usually don't add this database by default but I would go back and add it in a situation like this. In this case, your protein hits PF13411.9 ; MerR_1 ; MerR HTH family regulatory protein with 98% and 75% alignment. So in terms of assessing the quality of the hit I would say it is borderline (for PFam hits I do like nearly full length) but since this is just a specificity issue (I.e. there is plenty of evidence from your search that there is indeed an HTH in this protein) I would tell a student that, yes, this PFam hit is sufficient evidence to support the added "MerR like" term to the function. Note that some of the other proteins in your search do also mention the MerR like domain but you need to be careful and not take that as evidence at face value. Since HHPRED will happily report partial alignments, to use that evidence you would need to confirm that the MerR like HTH domain in the description is also in the region of the alignment. This can easily be done, it just usually takes a few clicks to dig into other databases and thus more time; whereas when looking for a good PFam hit, all the evidence you need is right there on the HHpred results page, so it is more of an expediency issue than a "quality of evidence" issue as to why I prefer using PFam. |
Posted in: Annotation → helix-turn-helix binding domain or protein?
| Link to this post | posted 19 Dec, 2022 23:20 | |
|---|---|
|
|
Ok I just tried this out and everything worked. I now have DNA Master running in a Windows 11 for ARM using the free "for personal use" version of VMWare Fusion 13 so this is a solution that does not require buying parallels. With all the registering, downloading and configuring it took about 2.5 hours. The video was very detailed and goes through all the steps. So if you are at all reticent I recommend watching the video linked about. If you want to just try I would add the following steps: step 4 add that after download you need to install homebrew on your mac, then use homebrew to install the qemu suite, then use qemu-img to convert the Windows 11 client download from a VHDX format to a VMDK format for use with VMWare. step 7. you will get stuck trying to get windows to start up as installation now requires an internet connection but you cannot install the network drivers and get internet until you isntall the VMtools which you cannot do until you start up Windows. So check out the video or search for on how to boot windows 11 without internet. Finally to get everything working once Windows fully boots, use Powershell in administrator to run the VMWare Tools from the virtual CD. I too have not been able to get file sharing or copy/paste set up. According to this page: https://communities.vmware.com/t5/VMware-Fusion-Discussions/Can-t-enable-the-shared-folders-with-Fusion-13-Player-free/td-p/2941943 and this page: https://communities.vmware.com/t5/VMware-Fusion-Discussions/Copy-Paste-file-sharing-between-Ventura-and-Windows-11-ARM-doesn/m-p/2940220 these features are not yet supported yet for M1 mac/Windows 11 for Arm. See suggested work arounds. |
Posted in: DNA Master → DNA Master on M1 Mac
| Link to this post | posted 29 Sep, 2022 15:37 | |
|---|---|
|
|
if it is asking for the user name that is:for the password use: |
Posted in: Bioinformatic Tools and Analyses → PhamNexus on SEA-VM
| Link to this post | posted 18 Aug, 2022 22:16 | |
|---|---|
|
|
That was very helpful. The HHPRED results will only be available for a short while and so I am posting the protein sequence of the example above so that anyone who wishes can redo the search. |