Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
cdshaffer posted in Whole phage starterator reports
Debbie Jacobs-Sera posted in frameshifts
uOttawaPHAGE posted in frameshifts
Dan Russell posted in Congrats to Steve Caruso and Beth Wilkes — 2025 ASM Outstanding Instructor Award, Honorable Mention
ACMPhageHunters posted in Clarification Question About HNH Endonuclease Function Determination in view of hits to the Ref Sequences
All posts created by cdshaffer
| Link to this post | posted 13 May, 2025 15:02 | |
|---|---|

|
yes. The genome is going to circularize after entry, at which point there is only one copy of the repeat within the genome. This means that annotations of the linear genome will always have the possibility of this kind of quirk. In addition, if you annotated a partial gene at the start of the genome it would cause all kinds of issues for the all the computational checks and make handling the genome much more labor intensive, so the best approach is as you suggest. Annotate the copy that is the full intact gene at the end and do not annotate the partial gene at the beginning. |
| Link to this post | posted 04 May, 2025 00:37 | |
|---|---|
|
|
Database 598 was released with 1 replacement phage to reflect a name spelling change. The whole starterator report for this phage has been added to collection found here just to have the correct spelling https://wustl.box.com/v/Actino-phage New Phage Released in 598: Schaffner |
Posted in: Starterator → Whole phage starterator reports
| Link to this post | posted 26 Apr, 2025 18:57 | |
|---|---|
|
|
Database 597 was released with 1 new phage. The whole starterator report for this phage has been added to collection found here: https://wustl.box.com/v/Actino-phage New Phage Released in 594: KillerQueen |
Posted in: Starterator → Whole phage starterator reports
| Link to this post | posted 19 Apr, 2025 22:54 | |
|---|---|
|
|
Database 596 was released with 4 new phage. The whole starterator reports for these phage have been added to collection found here: https://wustl.box.com/v/Actino-phage New Phage Released in 593: Keough Lilo27 MamaT & Skelbel |
Posted in: Starterator → Whole phage starterator reports
| Link to this post | posted 18 Apr, 2025 21:37 | |
|---|---|
|
|
You posted really good evidence in support of the presence of zinc finger domains which are often part of DNA binding domains. However in the Mol Microbiol. 2017 Aug;105(4) paper they note that the Fin protein actually has a protein:protein interaction with RNA polymerase. I only did a quick scan of the figures but it appars they did nuclear magnetic resonance chemical shift analysis of fin in the presence of B' to look for evidence for which amino acids are involved in this protein protein interaction. See fig 5C. In there it looks like the residues that are the most perturbed and therefor the most likely to be critical for binding are Gly 15 and Glu 45. So the question is are those residues conserved? if so then you might have an argument for an annotation but just having a zinc finger motif means the only annotation justifieed is "zinc finger doamin" and we usually do not annotate just domain. I think is especially valid in this case as most people, including myself, think of zinc finder domains as DNA binding domains but this paper say that Fin is a RNA polymerase binding not DNA. binding. So I would stick with NKF |
| Link to this post | posted 14 Apr, 2025 17:28 | |
|---|---|
|
|
As a follow-up. If you want to examine the predicted structures in 2D like aragorn you can use this site: https://rnacentral.org/r2dt Take any result from the tRNAscan struct file like this: Get rid of the "seq:" and "Str:" replace all > with ( and all the < with ) and then add a header line starting with > and adding a description Paste that into the web page and hit run to get a nice colorful 2D rendering, see attached. |
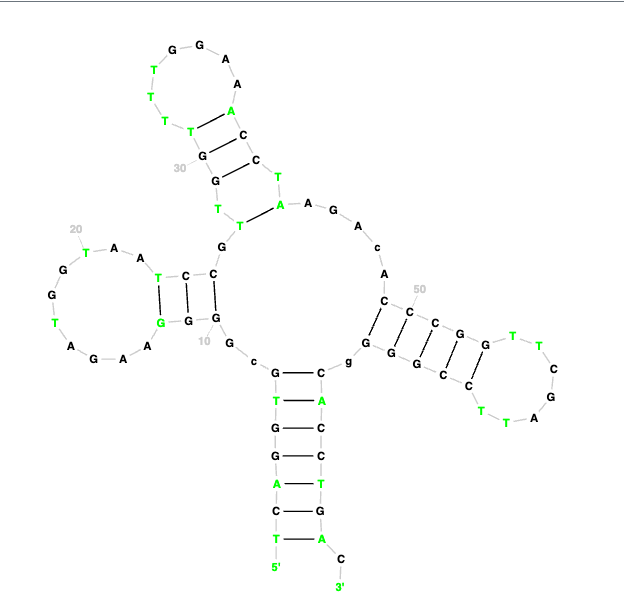 32Kb
32KbPosted in: Bioinformatic Tools and Analyses → tRNAscan-SE
| Link to this post | posted 14 Apr, 2025 16:13 | |
|---|---|
|
|
As was discussed in the Friday CAT session tRNAscan has been down for a few days. The good news is that the program is also available for download and can be run on the command line. With that in mind I just processed all 750 odd phages that are marked as draft with tRNAscan-se. If your phage is missing post a follow-up message to this thread. You can get your results at the following link: http://phages.wustl.edu/trnascan/ Your results will be a zip archive with 3 files. These files are named starting with the Phage name and include "info", "struct", and "table" as part of the file name. The "info" file was created to confirm that the phage was analyzed and has the exact details on run parameters and version (you will need the version number if you intend to publish). For documentation here: tRNAscan-se was run with parameters "-X 0 -d -B -I -D -q" and the exact version was 2.0.12. The "struct" file contains the predicted structure of the tRNA as printed by tRNAscan-se. The "table" file contains the results in a table format very similar to the table results you get from the web page. If your genome has NO predicted tRNA's the "struct" and "table" files will be there but will be empty. Just be aware that the command line version does not produce those nice structure predictions like aragorn. The structure results look something like the text below and must be manually parsed. For this, the struct files are best viewed using a monospace font like consolas or courier (not available in forum posts): |
Posted in: Bioinformatic Tools and Analyses → tRNAscan-SE
| Link to this post | posted 29 Mar, 2025 02:09 | |
|---|---|
|
|
Database 593 was released with 5 new phage. The whole starterator reports for these phage have been added to collection found here: https://wustl.box.com/v/Actino-phage New Phage Released in 593: FatCactus, Liberone, Neuvillette, RoseMarie, and Rossetti |
Posted in: Starterator → Whole phage starterator reports
| Link to this post | posted 22 Mar, 2025 00:56 | |
|---|---|
|
|
Database 592 was released with one new phage. The whole starterator report for this phage has been added to collection here: https://wustl.box.com/v/Actino-phage Phage Released in 592: WileyE |
Posted in: Starterator → Whole phage starterator reports
| Link to this post | posted 14 Mar, 2025 16:56 | |
|---|---|
|
|
Here is a simple example with super simple sequences:The %aligned is 100% because the entire query is found in the alignment the % coverage is 25% since only 3 bases of the 12 bases in the subject are in the alignment For % identity you get 100 % because 100% of the bases in the alignment match identically For DNA there is no %similar (the % similar is only used for amino acids alignment) but for a.a you just would count the fraction of alignment columns that are either identical or similar and divide by the length of the alignment. CCD is a database of protein domains (i.e. small parts of proteins seen widely) think things like zinc finger or ATP binding domain. Thus, for interpretation of CCD hits hits you care most about the % coverage and % similar the % aligned is mostly irrelevant. |