Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
Dan Russell posted in Congrats to Steve Caruso and Beth Wilkes — 2025 ASM Outstanding Instructor Award, Honorable Mention
ACMPhageHunters posted in Clarification Question About HNH Endonuclease Function Determination in view of hits to the Ref Sequences
cdshaffer posted in Clarification Question About HNH Endonuclease Function Determination in view of hits to the Ref Sequences
All posts created by cdshaffer
| Link to this post | posted 11 Nov, 2021 22:55 | |
|---|---|

|
OK follow up on the issue with pham 56633. As I said before I had already found that the issue was with phage ISF9 gene 29. It turns out that phage is one of the "Added phage" which don't come from any of the Pittsburg programs but was a phage isolated from Microbacterium oxydans in Iran and published in genbank. It turns out this sequence has two N bases in the version of the sequence in Actino_Draft and these N bases confused Starterator and caused it to crash when it was counting bases to find the start and stop codons. So this bug should not be a problem for phage that we publish since Dan is always careful to check the sequences for N's but it could be an ongoing issue for these phage that get added. Not sure exactly how to deal with these phage in the long run but for now please continue to post if you find a missing pham report. |
Posted in: Starterator → Pham not found in Starterator
| Link to this post | posted 11 Nov, 2021 20:24 | |
|---|---|
|
|
Very cool, I think it could be fun to set it up so students could do their own sequencing on a nanopore sometime late in the first semester. These genomes are so small I think we could get enough data even on the smallest (i.e. cheapest) of the nanopore sequencers. Which did you use? Were you using the standard Minion or one of the smaller flongles? OR was this outsourced on a GridION? Thanks for being the initial test subject. The only comment I have is that I thought there was a certain rate in which the pore will pick up and start sequencing the second strand pretty quickly after the 1st. Does your single long 115k read look like an inverted repeat? This is what I would anticipate if you were reading the second strand after pulling the first strand through the pore. Have you tried assembly yet? Do you get a single contig of the size expected for an AW phage? If so I would think you get an estimate of the quality of the genome assembly by just running the draft assembly through auto-annotation. We have such a strong expectation of "tight pack" genes and since many assembly/sequencing errors would disrupt genes, I would think an auto-annotation of that preliminary assembly could provide a decent estimate of the quality and if there was a need for illumina polishing or not. |
Posted in: Sequencing, Assembling, and Finishing Genomes → Nanopore
| Link to this post | posted 09 Nov, 2021 21:24 | |
|---|---|
|
|
OK preliminary analysis suggests this is some kind of error in start codon annotations in phage ISF9 gene 29. This is a non-SEA phage from genbank that was added to the Actino_Draft database. The annotated start for this gene in the Actino_Draft is not a valid start codon once it is analyzed by Starterator. So it could be a bug in Starterator or a data entry error in Actino_Draft database. Determining that will take time, but in the mean time I just hand edited my local copy of the database to remove the problematic gene from pham 56633. I then ran the starterator analysis with all members of the pham except ISF9_29. The report should now be available but you will want to download the file for later use as it is likely to disappear again with the next database update, as I am not sure how long it will take to track down the exact issue. For documentation purposes this link should work for the next 3-4 months: http://phages.wustl.edu/438/Pham56633Report.pdf |
Posted in: Starterator → Pham not found in Starterator
| Link to this post | posted 09 Nov, 2021 17:53 | |
|---|---|
|
|
Wow thanks for this, you are correct that pham is missing. You have actually located a bug in Starterator, which crashes when you try to analyze that pham. This is excellent, as we find and fix these bugs the program works better and better. I did a quick check and I found the general location of the problematic pham in the analysis but will need time to dig deeper into why it is happening. I will post more info once I get this bug squashed. Thanks for posting. |
Posted in: Starterator → Pham not found in Starterator
| Link to this post | posted 03 Nov, 2021 16:25 | |
|---|---|
|
|
As usual we are in agreement. I too think it is a really strong candidate but I was just not going to call without something more in support. |
Posted in: Frameshifts and Introns → TAC frameshift in singleton
| Link to this post | posted 01 Nov, 2021 19:58 | |
|---|---|
|
|
OK, I have the situation in phage onionknight (a singleton) looking for a tail assembly chaperone and looking for the slip. The attached shows the region which is the only sequence that matches at all with any of the published slippery sequences. However, it is by no means an exact match. See pic, I have highlighted the putative slippery sequence in blue and the two translation frames in red. This would be a typical -1. This slip would be about 20 amino acids upstream of the stop codon in the short form so not an unreasonable location, it would just be nice to get a consensus with other annotators if this match to published slippery sequences is close enough to annotate or should we just leave it as two separate ORF's. |
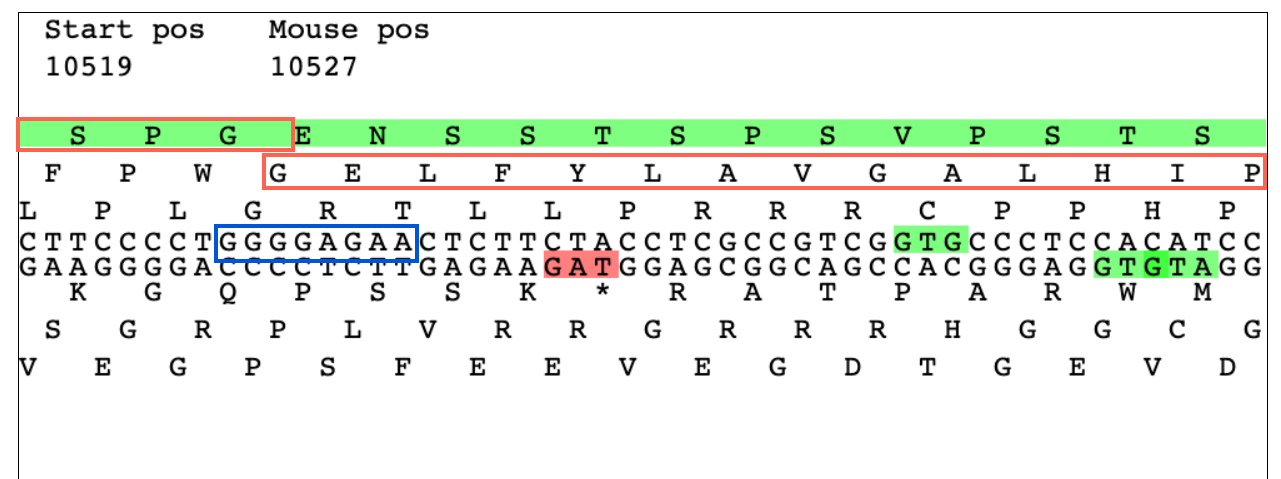 126Kb
126KbPosted in: Frameshifts and Introns → TAC frameshift in singleton
| Link to this post | posted 16 Sep, 2021 20:51 | |
|---|---|
|
|
I will add my experience as another datapoint to help with troubleshooting: I too am also getting glimmer results but not GeneMark results, and I have confirmed the settings as displayed in Debbies attached picture. Very strange that one predictor would work and not the other for some users and not others. |
Posted in: DNA Master → Auto-annotation fix for fall 2017 and later
| Link to this post | posted 13 Sep, 2021 18:04 | |
|---|---|
|
|
That looks like a very low level issue with the disk device drivers. Very difficult to troubleshoot without a lot more details or direct access to your machine. I would try deleting this machine and rebuilding. You could also try the newer SEA 2020 VM. The good thing about that 2020 VM is it has the new pdm_utils installed. I don't know if I am allowed to post the link to the VM disk image in the open. Feel free to email and I can send you the link. |
| Link to this post | posted 20 Aug, 2021 00:55 | |
|---|---|
|
|
Nancy, These discrepancies typically happen due to the database updates coming out at different times for the different sites. things can often slow down a bit in the summer with vacations and the like as well. You can use these links if you want to see which version of the database is being used at each site: phagesdb see: http://databases.hatfull.org/Actino_Draft/Actino_Draft.version starterator see: http://phages.wustl.edu/starterator/database.version pecaan: look on any "pham maps" page just above the map phamerator.org: open the pull down menu in the top left Currently Starterator and Phamerator are behind on 424 and phagesdb is on 425, so you want to use the phamerator number to get to the starterator report. The instructions on the various ways to get to the starterator report if the one from phagesdb is not working can be found here: https://seaphagesbioinformatics.helpdocsonline.com/article-37 In this particular case, since you know you want pham 73206 you can just type the address out manually (or copy/paste this address ). http://phages.wustl.edu/starterator/Pham73206Report.pdf That page has the report on Ganymede_Draft gene 1. |
Posted in: Starterator → Pham not found in Starterator
| Link to this post | posted 04 Aug, 2021 21:56 | |
|---|---|
|
|
newer browsers have dropped support for the FTP: protocol. There was a while when firefox was working even though chrome was not, so try firefox. You can also try Dan's instructions here: https://seaphages.org/forums/post/8693/ Although if you read through the rest of that discussion, it appears lots of people are having trouble with new installs even though updates on already installed versions are still working. But I cannot tell if it is failing for everyone or just some new installs. Good luck. |
Posted in: DNA Master → Problem installing DNA Master on Windows 10