Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
New Features in PECAAN
| Link to this post | posted 20 Mar, 2017 20:01 | |
|---|---|

|
Today I noted that there are 914 registered users of PECAAN and 230 phages in the PECAAN database. Since we have recently added a couple of new features that I have found especially useful in checking my student's annotations, I thought that I would highlight these in a post here just in case you had not discovered them yet. PECAAN now used both Aragorn and tRNA Scan SE to call tRNAs and tmRNAs. These pages are accessible through the top menu. By moving the vertical slider on the top sequence you can adjust the start and stop sites. Both Aragorn (red region) and tRNA Scan SE (blue region) calls are displayed on the top sequence map. Evidence and pictures of structures are shown for each method. An "tRNA Included" checkbox allows you to include (checked) or not include (unchecked) the tRNA. Edit and review logs for users are kept similar to the Genes log. Reports associated with tRNAs can be exported from the "Export" menu. The "Export CDS Full Annotation" report now includes the tRNAs and tmRNAs so that you do not need to edit the report before importing into DNA Master. A recent feature, that I am delighted with, is the "Pham Maps" page, which is available in the top menu. This page allows you to see a map of the latest edited genome features in a map of the Phams (yes, we use the standard pham colors). The map includes genes as colored boxes, if there is a pham number available, and tRNAs as + symbols. You can select one genome, from the cluster to which your working genome belongs, to compare to in a second line in the map. If you are displaying a gene and want to look at the map, clicking on the Pham Maps menu will take you to the map location containing the gene. Since I have multiple displays, I simply right click on the Pham Maps menu and either display the map in a separate tab or preferably in a separate window that I move to a separate monitor. I have found the Pham Map to be a super tools for reviewing student annotations because it is easy to see their gene calls and annotations in comparison to a published phage. I can see the pham number, start, stop and function for each gene I mouse over each gene in the map, so it is easy to comparatively check both the location and annotations from this one display. If you go to the Gene window and make changes for the "start" or "include" features and then go to the Pham Map the changes will be immediately reflected as it is refreshed. The great thing about Pham Maps is that you can change the Pham database phage that you are comparing your genome with! This allows you to compare different phages to your genome as you move along the genome during annotation and checking. Finally, we have made some changes in the User and Phages pages that are found under the Admin menu. These changes should make it easier to navigate through and edit within each of these pages. If you have questions or problems with PECAAN, Please email me at: claire.rinehart@wku.edu or call 270-745-6892 (M-F) 9-5 CT. |
| Link to this post | posted 21 Jun, 2017 13:13 | |
|---|---|

|
How do I get the Staterator start to appear in the list for a gene? I can see Glimmer and GeneMark, but the Starterator field is blank. I can see the pham number. Also on the phage entry page, for the Starterator data file - is that the Starterator report pdf? Thanks! Janine |
| Link to this post | posted 21 Jun, 2017 19:41 | |
|---|---|
|
|
The Starterator field in PECAAN is a legacy field to accommodate manual entry from an edited Starterator report that we don't encourage use of anymore. The best way to view the Starterator information is by clicking on the Pham link. This loads a Starterator summary that is very useful. After clicking on the Pham link, scroll down to the text: "Summary of Final Annotations (Info on gene starts based on numbers in diagram): The start number called the most often in the published annotations is start number" X, where X is the most published start number, as displayed in the tracks (map at the top). This is the number that Staterator recommends. (Helpful navigation hint: Using the Find command for your machine will help you navigate to find information for your specific phage. Type your phage name into the Find field (activated by command F on the Mac) and press the keys (command G on the Mac) to find the next instance of your phage name in the text.} Next, there are three classifications for genes: Genes that call this "Most Annotated" start. Genes that have the "Most Annotated" start but do not call it. Genes that do not have the "Most Annotated" start:Genes that do not have the "Most Annotated" start. Use the navigation hint to jump to your gene and note which of the three classification that it fall into. The next section of the Starterator report lists the called genes under each start number. This can be useful if there are only drafts in the pham or if your gene does not have the most recommended Starterator site. This can give you a feel of how represented the called start site is in the context of all genes in the pham. The final section lists the Gene Information, which the list of (start numbers, start positions). This is where you can go to translate the start numbers into their actual genome positions. The first line shows the start that was called, but most useful are the next lines that list all of the start locations and their corresponding Starterator start number. This is where you can correlate the start numbers with your possible translation start locations in the gene. Hope that this helps a little. Claire |
| Link to this post | posted 21 Jun, 2017 19:48 | |
|---|---|
|
|
It helps a lot! Thank you, Claire, for this extremely helpful software. |
| Link to this post | posted 31 Jul, 2017 15:50 | |
|---|---|
|
|
Just a note to update you on some of the latest features added to PECAAN. Whenever you begin to type a function into PECAAN it will show you all of the matching functions that are found in the "approved function list". An easy way to take advantage of this feature is to type in all of part of the root function and then select the appropriate function from the drop-down list. With the initiative to only enter "approved functions" we have also included an indicator of membership in the "approved function list" in the gene number selection drop-down at the top of the Genes window. If a function is on the "approved function list" then a green checkmark will appear before the gene number. If a function is not on the "approved function list" then a red x will appear before the gene number. This should help in reviewing whether the functions, that have been entered for the genes, have the correct description. We have also added a Function option, under the Admin menu item, that allows SuperAdmin users to modify and update the approved function list. |
| Link to this post | posted 22 Jan, 2018 16:48 | |
|---|---|

|
Hi Claire, Does it take a while for starterator and pham data to appear on the site? Over break I annotated a large number of genomes on Pecaan and don't remember it taking a long time for this data to appear. It has been at least a week since I entered five new genomes and the HHPRED and BlastP data is there but not the starterator/Pham data. Thanks! Sally |
| Link to this post | posted 22 Jan, 2018 18:17 | |
|---|---|
|
|
Sally, I assume that you are referring to Sansa, Aminay, Steamy, Acolyte, and LastResort. I checked on the web Phamerator and could not find them therefore I wonder if they have been Phamerated. PECAAN talks to PITT and St. Louis to pull Phamerator and Starterator information respectively. If a phage hasn't been Phamerated then I don't think that it will show up in the Pitt database. Thanks, Claire |
| Link to this post | posted 23 May, 2018 17:29 | |
|---|---|
|
|
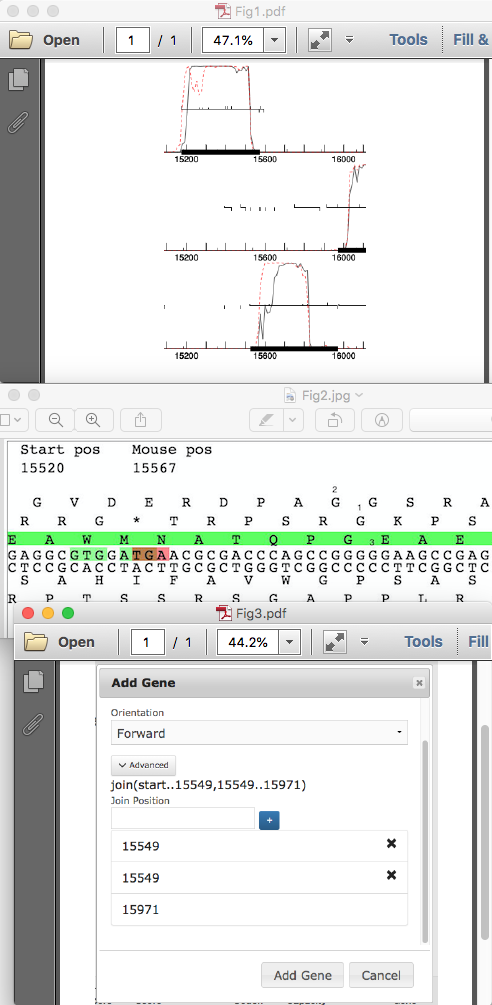
Ruth Plymale recently asked how to annotate a frameshift in PECAAN. Since this is a new feature in PECAAN, I thought that I would post the answer here also. Since I am currently working on AugsMagnumOpus I will demo how to find and record frameshifts with this phage. First a quick look at the GeneMark coding capacity plot shows the coding capacity for the two parts of the longer frameshifted gene: 15175 – 15573 and 15520-15971. (Fig. 1) Notice that the left half is in the top forward reading frame and overlaps with the right half that is coded for in the bottom forward reading frame. If you go to the Sequence menu in PECAAN and scroll down you will see the six-frame translation. The top three are forward translation and the bottom three are reverse. All potential Start (green) and Stop (red) codons are displayed in the Top and Bottom DNA strands. The amino acid in each translation is oriented over the first base of the triplet codon that codes for it. If you type 15520 into the Seek position: box in the Sequence window and press return, the left side of the window will scroll 15520 (Fig 2). Notice that the green gene highlight is in the bottom forward translation and corresponds to the left gene. Recall that in GeneMark, this was in the top forward frame. So, the correlation in frames between PECAAN Six Frame and GeneMark is as follows: PECAAN Six Frame……….GeneMark Forward Top…………..Forward Bottom Forward Middle………..Forward Middle Forward Bottom………..Forward Top Therefore, we will expect the frame shift to move from the bottom reading frame in the PECAAN Six Frame figure to the top reading frame since in GeneMark the left half was in the top reading frame and the right half in the bottom reading frame. If we look for a potentially slippery spot in the Forward DNA strand we see in the middle of Figure 2 there is a stretch of five Gs that correspond to PGE in the bottom translation and AGGS in the top frame translation. During the frameshift the tRNA with the Glycine under label 1 shifts back one base to correspond with the Glycine under label 2 in Figure 2. Therefore, the Guanine under label 3 in Figure 2 is both the last base used in translation of the bottom reading frame and becomes the first base in continuing translation of the top reading frame. To accurately find the position of the G under label 3 in Figure 2, take and drag the figure to the left until the G, under label 3, is positioned as the first base at the left of the window and then use the number under Start pos as its proper location which is 15549. Using the Mouse pos can sometimes be inaccurate and should be used to get general locations. Therefore the two parts of the frameshifted gene are then defined by the ranges 15175 .. 15549 and 15549 .. 15971. Now, return to the Genes window by clicking on Genes in the top menu. Click on the top Add Gene button and the Add Gene window will pop-up. Enter the Gene Stop as 15971 and ensure that the Forward Orientation is displayed. Click on the Advanced button and another field will appear for you to enter the beginning of the right half of the frameshifted gene. Enter 15549 and press the blue + button. Another field will appear that will allow you to enter the right end of the left half of the frameshifted gene which is also 15549 in this case. Again, click the blue + button and you will see the complete entry shown in Figure 3. At this point click the Add Gene button at the bottom of the window and the gene will be added and you will see join(start..15549,15549..15971) as the gene range for the new frame shifted gene. One last thing. You will need to go to the original gene representing the right half of the frame shifted gene and uncheck the Gene Included box so that you don’t have it included in the genome. You should see it designated as (Not Included) when you look at the gene selection drop-down menu. |
 163Kb
163Kb| Link to this post | posted 24 Jul, 2018 15:50 | |
|---|---|

|
Hi Claire, I have used Pecaan to annotate Sonali. This is the first time I have used it and I am now exporting the files. Is there a way to place the Function in the Function box for the complete annotations? Thank you, Tammy |
| Link to this post | posted 24 Jul, 2018 17:00 | |
|---|---|
|
|
Tammy, We currently do not place the function into the function box in our DNA Master Full Annotation export. Welkin just pointed out to me that the online guide tells how to generate minimal files from the complete notes when functions are recorded in the function field. We will work on adding this feature to the complete file export. Meanwhile, you can use the Export CDS Function button on the export page of PECAAN to export a file that can be copy/pasted into the DNA Master Documentation page which will parse the functions into the notes field. These minimal functions can then be copied en masse into the product or function fields in DNA Master by clicking on the right hand triangle in the Notes field. Thanks, Claire |