Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
Dan Russell posted in Congrats to Steve Caruso and Beth Wilkes — 2025 ASM Outstanding Instructor Award, Honorable Mention
ACMPhageHunters posted in Clarification Question About HNH Endonuclease Function Determination in view of hits to the Ref Sequences
cdshaffer posted in Clarification Question About HNH Endonuclease Function Determination in view of hits to the Ref Sequences
All posts created by Pollenz
| Link to this post | posted 08 Jun, 2023 00:22 | |
|---|---|

|
L5 is listed as GGGGGAA with two A's both in the paper and in the nice table. Its the same issue with using GGGAAA, that is formally listed as being GGGAAAA with 4 A's, not three.
RS Pollenz
|
Posted in: Cluster EA Annotation Tips → frame shift
| Link to this post | posted 07 Jun, 2023 20:26 | |
|---|---|
|
|
Review of EA4 phages at the 2023 faculty meeting shows a sequence of CGGGGGAc that has been annotated many times as the slippery sequence. While this has the 5Gs, it does not match exactly to the sequences listed in the Guide as they have the sequence CGGGGGCG or GGGGGAA. While compelling, are we calling sequences that are "almost" identical?
RS Pollenz
|
Posted in: Cluster EA Annotation Tips → frame shift
| Link to this post | posted 07 Jun, 2023 13:24 | |
|---|---|
|
|
Review of this cluster at the 2023 Faculty meeting revealed many phages that have been annotated with a slippery sequence that DO NOT meet the sequences listed as validated in the Bioinformatics guide: https://seaphagesbioinformatics.helpdocsonline.com/article-54 These include CGGGGCG (should be CGGGGGCG: note there are 5 Gs) and GGGAAA (the correct sequence is GGGAAAG, GGGAAAT or GGGAAAA, note there are FOUR nucleotides after the GGG, not three). It is essential to assure that when you call the slippery, it matches to the list in the phages guide, otherwise we have a big mess and it propagates miscalls. As more wet lab data is developed, the list is updated, so this should be checked each year you are annotating.
RS Pollenz
|
Posted in: Cluster EG Annotation Tips → TAC Annotations
| Link to this post | posted 25 May, 2023 12:25 | |
|---|---|
|
|
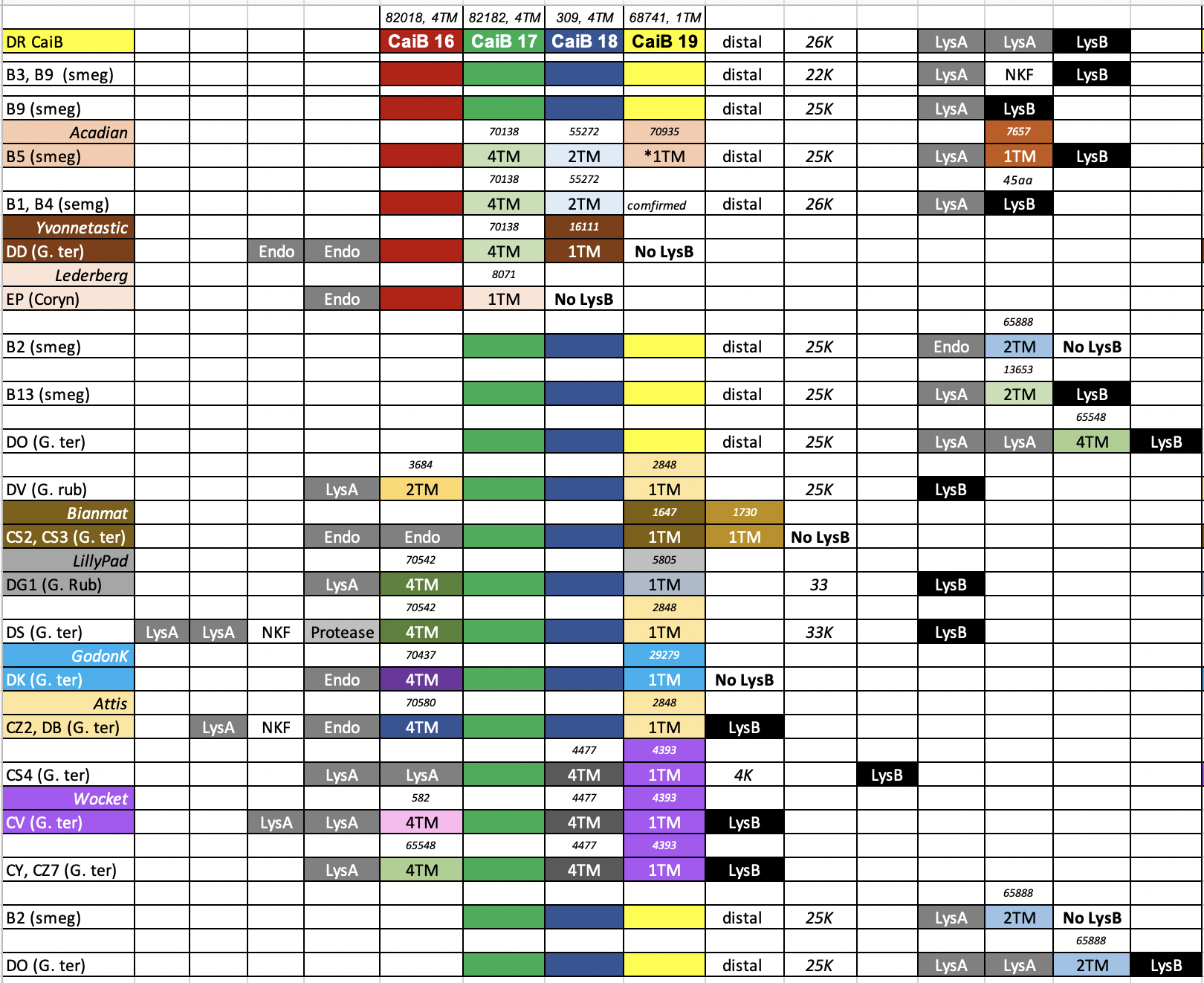
We have use a Bioinformatic approach to identify several phage clusters that have a distal holin-like gene cassette that are not associated with the lysins. This type of organization can be observed in the B1, B2, B3, B5, B9, DR and DO phages. Most of the cassettes have four different TM genes with a 4-4-4-1 configuration and in many cases all these genes have a 4bp overlap to each other. We have bioinformatically looked at all of the pham members for all of these distal holin-like genes and CAN FIND THEM DIRECTLY DOWNSTREAM of lysins in DD, EP, DV, DG1, DK CZ2, DB, CS4, CV, CY and CZ7. This clearly validates that they are likely functioning in lysis. I have enclosed an colorful analysis based on the 4 genes in the DR Phage CaiB. Each color represents a protein in the same pham. In the case of your B1 phages, note that phages in the DD cluster have both 4TM genes directly downstream of a split lysin followed by a 1TM gene. Based on our wet lab recent work, it is likely that ALL of the TM genes are holins (including the 1TM that may be the most important one. DATA IN PREP but presented at 2023 Phages Conference). Note that the majority of these multi-TM gene cassettes have a terminal 1TM gene. See our PLOSONE paper as well for a comprehensive analysis of these types of TM cassettes. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0276603
RS Pollenz
|
 1011Kb
1011KbPosted in: Cluster B Annotation Tips → Holin
| Link to this post | posted 11 Feb, 2023 22:52 | |
|---|---|
|
|
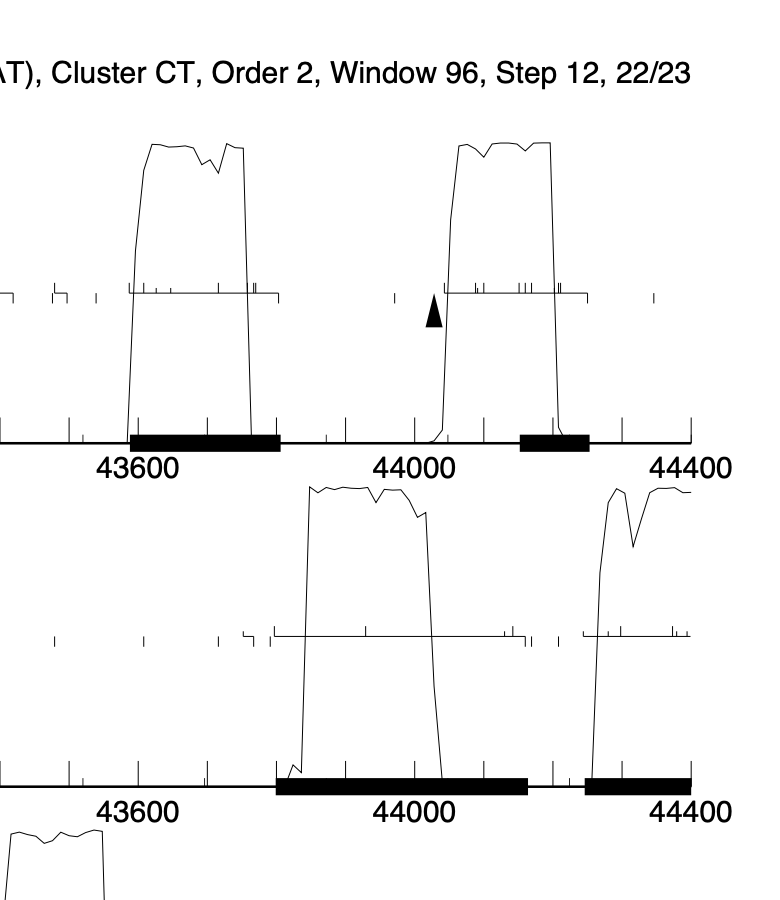
We are annotating phage Azira. It has high identity to survivors. In Azira, it is clear that gene #66 loses coding potential 1/2 way through the ORF, most likely due to losing a stop site. Gene #67 has coding potential across the entire ORF and this gene in most PHAM members is annotated with the longer ORF. Calling the LORF results in a 121bp overlap to gene #66 and this is what has been annotated for Survivors. All other phages that have this gene, do NOT have long overlaps to the previous gene as they all have smaller genes that precede it. Its obvious that Azira gp67 should be annotated to include the full ORF, especially since gp66 has no CP right at the point that gp67 starts, just as Survivors, but this highly violates the gene overlap rules. Anyone have any thoughts on this. I enclosed the Genmark map and you can see the situation, Azira gp67 is the gene starting at 44,000. As an added intrigue to this situation, there is a start site at 44161 for gp67 with a -4bp overlap to the end of gp66. Calling this results in a 93bp ORF and 31aa protein. Also violations of the general rule that ORFS are usually >120bp….
RS Pollenz
|
 75Kb
75KbPosted in: Cluster CT Annotations Tips → BIG GENE OVERLAP
| Link to this post | posted 21 Dec, 2022 21:01 | |
|---|---|
|
|
Great work on this question. The ribbon-helix-helix is a DNA binding motif found in many proteins. It is typified by a B-sheet of 6-10 amino acids followed by a short spacer and two alpha helices. It can be found in many locations within a primary sequence of a protein, but must contain all THREE domains. In HHpred the alpha helices are noted by Hh and beta sheets by Ee in the ss_pred line of the alignments. Capital letters indicate high confidence in the secondary structure (ss) prediction. See PDB 6SBW_A for a good schematic of this type of ribbon HH domain (enclosed schematic). Note that Usavi70 has alignments to the subjects with Q1 to all hits. Amino acid 1 is the start of a helix and there is also a second helix in Usavi70, but the Usavi70 has NO beta sheet that precedes the hits compared to the queries. See that the deeper analysis of the PDB queries show that they are all aligning well past amino acid 1 and all have a 7-9 amino acid beta sheet that precedes the helix 1 that is aligned to Usavi70. Thus, the ribbon helix helix IS NOT supported for Usavi70 and should not be called. NCBI conserved domains also does not support either an HTH or DNA binding call as well. So best to leave a hypothetical NKF since all components of the domain are not present. Hope this is helpful
RS Pollenz
|
 485Kb
485Kb| Link to this post | posted 24 May, 2022 18:44 | |
|---|---|
|
|
The DR Cluster phages appear to have a 3-5 gene transposable operon with -4bp overlaps at the 5' end of the genome prior to the terminase gene. There are several variations of the operon across all the phages with some shared genes and ouliers like Evaa and Sour that are clearly divergent from the majority of cluster members. Most genes within these "operons" have HHpred hits, but specific functional calls to some of the genes can not be made as the hits do not match to those on the Official genes list. These operons appears to encode genes involved in nucleotide modification and may be involved in phage defense against restriction as noted in Nucleic Acids Research, 2013, Vol. 41, No. 16 7635–7655. Note that the 5 gene operons contains a possible adenylate kinase gene that is very tricky and requires deep review of the HHpred hits as finding the phosphate-binding loop (P-loop) that is a conserved sequence motif found in mononucleotide-binding proteins/kinases and DOES NOT specify a specific kinase (Ex. compare the hits of CaiB_4 to LittleMunchkin_4). Adenylate kinases do have crystal structures in HHpred (2C95_B, 2BWJ_B, 3UMF_A) and cd hits in NCBI so there should be evidence beyond the P-loop to make the call. See also the notes on Gene 1 from Cluster B.
RS Pollenz
|
Posted in: Cluster DR Annotation Tips → Metabolic Operon
| Link to this post | posted 20 May, 2022 19:20 | |
|---|---|
|
|
Many of the G1 phages are highly conserved at the nucleotide level in the region between the last reverse gene (immunity repressor) and first forward gene (cro). The operator site can be used to guide the annotation and is 5'-CGACATATGTCG-3'. Note that wet lab data supports the identification of the putative -10 and -35 binding sites and this orients the location of the CORRECT start codon (see the paper referenced above). In some cases, the DNA master RBS scores for the correct start site ARE NOT VERY GOOD and the start may not have been chosen by Genemark or Glimmer. However the wet lab data from BPs should be used to annotate the proper starts. Note that this results in smaller protein products, but DOES NOT reduce HHpred hits to the relevant PDBs or truncate N-terminal amino acid sequences. Remember that the intergenic region when moving from a reverse to forward gene MUST contain a sufficient gap (usually of at least 50bp) to accommodate these regulatory regions for transcription of these genes.
RS Pollenz
|
Posted in: Cluster G Annotation Tips → integration cassette
| Link to this post | posted 20 May, 2022 17:06 | |
|---|---|
|
|
I have done some analysis of RecE while annotating G1 phage ShaboiShabazz gp42. First, the PDB hit to 3H4R_A are to an E. coli exodeoxyribonuclease VIII, this is the putative RecE gene and has no connection to Cas4. Second, the publication regarding this RecE gene has identified several “conserved” domains and the genes within the PHAM that includes ShaboiShabazz gp42 contain 4 of these domains with near 100% conservation of not only the key amino acids, but also the secondary structures (B sheets and alpha helices). Third, RecE (exodeoxyribonuclease VIII) function in concert with the RecT. RecE binds to free double-stranded DNA(dsDNA) ends and processively digests the 50-endedstrand to form 50-mononucleotides and a 30-over-hang that is a substrate for single strand annealing promoted by RecT (2009: Structure 17, 690–702). ShaboiShabazz gp43 hits to PF03837.17: RecT family. Its key to note that RecT functions in BOTH RecA-dependent and RecA-independent DNA recombination pathways, so having a hit to this Pfam is not specific to Cas4 and I’m not sure how these connections were even made. **SEE THE POSTED ANALYSIS OF gp42 showing the clear hits to RecE. For Pfam 12684.10: This entry represent a PD-(D/E)XK endonuclease-like domain superfamily. PD-(D/E)XK nucleases constitute a large and highly diverse superfamily of enzymes that display little sequence similarity. However, they share a common core fold and a few critical active site residues and ARE NOT specific to Cas4 either. Makes sense for RecE to hit to this Pfam based on overall function. Thus, hits to 3H4R_A SHOULD CALL RecE and having a downstream RecT also confirm that calling both genes is valid. Thus: ShaboiShabazz gp42 is RecE-like exonuclease and gp43 is RecT-like ssDNA binding protein.
RS Pollenz
|
 1.77Mb
1.77MbPosted in: Functional Annotation → Refining the call for Cas4 family exonuclease vs. RecE-like exonuclease
| Link to this post | posted 09 May, 2022 16:00 | |
|---|---|
|
|
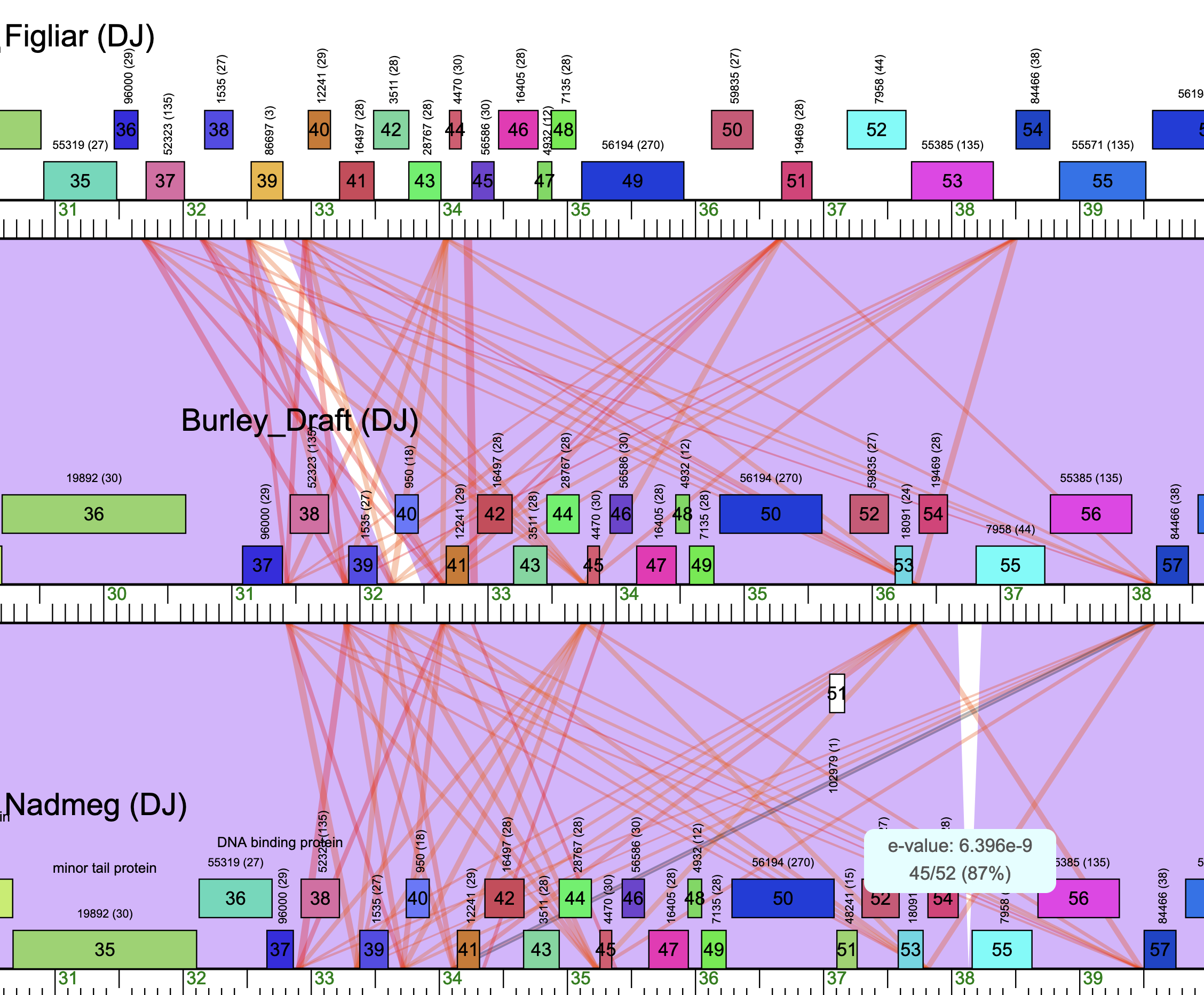
In the DJ phages (and also in many of the Gordonia clusters) there is evidence for numerous intergenic 50-75bp repeated sequence are are located directly upstearm of the ATG start sites for 7-10 genes in the middle of the genome, (in the DJ the area is just upstream of the tape measure gene). Note that these areas are easily identified when looking at the Phamerator maps as solid lines that diverge to numerous areas and hit the genome UPSTREAM of numerous genes. The nature of the sequence can be found using the SCAN function in DNA master and also using programs like CLUSTAL OMEGA to identify the alignments. They key is that although gaps of 50-75bp between genes are atypical, in some cases there are start sites that can be found within these regions that create longer ORFS. These longer starts should not be called and its important to assess both the Glimmer and Genmark calls as usually they are the correct start for these types of genes. In some cases the longer starts have been annotated and this creates confusion.
RS Pollenz
|
 1.31Mb
1.31MbPosted in: Cluster DJ Annotation Tips → Intergenic Repeated Sequences