Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by lhughes
| Link to this post | posted 26 Jan, 2024 14:51 | |
|---|---|

|
Fred - my understanding is that each program determines how to label the different frames independently, so there is no "standard" for which is going to be -1, etc. You need to determine which aligns with which each time you compare the data from two different programs to make sure you are comparing apples to apples. Lee |
| Link to this post | posted 12 Jan, 2024 21:09 | |
|---|---|
|
|
Congratulations on your retirement, David! It has been wonderful to have the opportunity to interact with you as the SEA has grown. Thank you for all you have done to help make this possible. All the best, Lee Hughes |
Posted in: General Message Board → A Message from David Asai
| Link to this post | posted 12 Jun, 2023 13:39 | |
|---|---|
|
|
Hi Steve, If you can't get the DNAM workaround from Kristen to work for your class, I'm happy to share how we use PECAAN in our classes (slides and other resources). We use PECAAN first in our annotation classes. Lee |
Posted in: PECAAN → Teaching with PECAAN
| Link to this post | posted 07 Jun, 2023 16:53 | |
|---|---|
|
|
Kathleen, Did you ever get an answer to this question? We just got sequence back on a new BM and will be annotating it in the near future, so I am interested in what you found out. If you haven't gotten an answer yet, it might be worth emailing Chris Shaffer for more information. Lee |
| Link to this post | posted 09 May, 2023 13:46 | |
|---|---|
|
|
Thanks, Debbie. Good to know it's not just me, and thanks for the thoughts on a workaround. Lee |
Posted in: DNA Master → DNA Master Fetch by Accession error
| Link to this post | posted 08 May, 2023 21:00 | |
|---|---|
|
|
I have been getting errors when trying to download some genomes from GenBank, but not all. I'm attaching an image of the error I received today trying to download Streptomyces phage Andris. I'm not sure if this is part of the problem or not, but all the genomes that have given me this error have been more recent (I've verified that they are active in GenBank) and seemed to have an accession number starting with the letter "O". Lee |
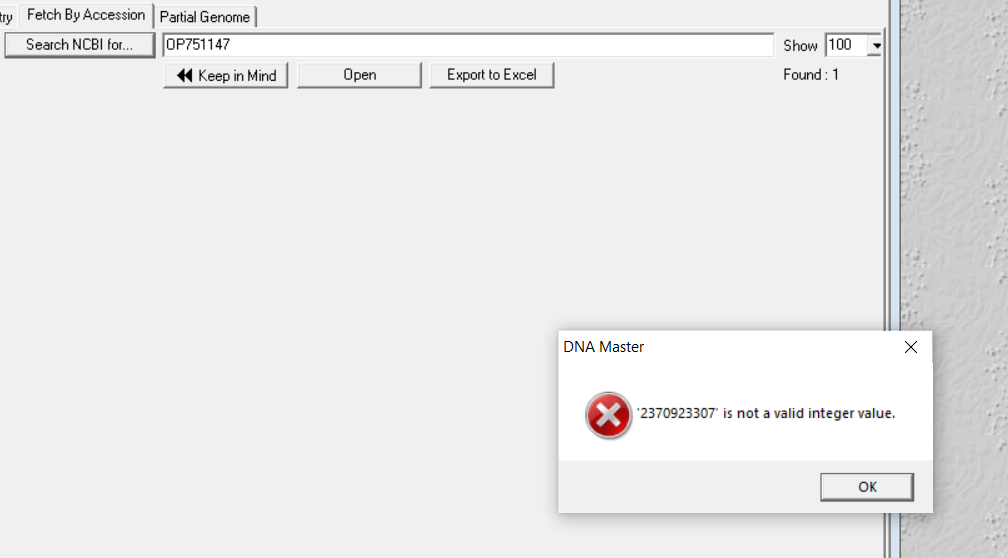 87Kb
87KbPosted in: DNA Master → DNA Master Fetch by Accession error
| Link to this post | posted 19 Apr, 2023 20:31 | |
|---|---|
|
|
Thanks, Debbie. I noticed all of our (UNT) more recent genomes that have a member of this Pham have used the "phosphatase" function so that makes the most sense given that we would need more evidence in order to call the more specific function. Lee |
Posted in: Annotation → 5’nucleotidase v. phosphatase
| Link to this post | posted 18 Apr, 2023 14:08 | |
|---|---|
|
|
Can I add a wrinkle to your question (I also have a phage that we are currently annotating that has a member of this Phamily)? In the Official Functions list it gives ClubL144 as the example for the 5'nucleotidase function but when I look it up on PhagesDB that gene has a function of "phosphatase" showing on the Pham page. Lee |
Posted in: Annotation → 5’nucleotidase v. phosphatase
| Link to this post | posted 12 Jul, 2022 13:29 | |
|---|---|
|
|
Thanks for those insights, Chris. We haven't annotated a cluster BM phage yet, but I like how this case shows how important context can be when making these decisions. |
| Link to this post | posted 19 Nov, 2021 16:19 | |
|---|---|
|
|
Awesome! Thanks, Debbie. Lee |
Posted in: tRNAs → Tomas tRNA error