Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by afreise
| Link to this post | posted 09 Mar, 2022 17:50 | |
|---|---|

|
Oh, I just realized this data pull does include draft genomes - so my first question is answered. |
Posted in: Bioinformatic Tools and Analyses → Difficulty pulling large-scale data for batch GCS analysis using Pope/Mavrich 2017 scripts
| Link to this post | posted 09 Mar, 2022 17:32 | |
|---|---|
|
|
WOW - thank you SO MUCH, Christian! It worked perfectly! Definitely easier than what I was attempting.  A couple more questions, if/when you have a moment - -Is there a way to do the same analysis but include draft genomes? I want to do a preliminary analysis including drafts just to see where some of the newer AZ phages fall that haven't been manually annotated yet (not for publication, but just out of curiosity) -If I wanted to compare phages from multiple clusters, is that possible using an adjustment to the above script? For example, we are interested in including singleton phage BaileyBlu to the AZ phages, and/or other clusters such as EH. |
Posted in: Bioinformatic Tools and Analyses → Difficulty pulling large-scale data for batch GCS analysis using Pope/Mavrich 2017 scripts
| Link to this post | posted 09 Mar, 2022 15:56 | |
|---|---|
|
|
Update: I've generated the files successfully! I think now I just need to pare down in excel to the only phages I want to compare. Right now gene.csv has all 4000+ phages and genes and I think my computer will explode if I run the GCS script on all of them. Even though I've only listed my subset of AZ phages in genome.csv, I should still need to pare down gene.csv to those phages too, right? |
Posted in: Bioinformatic Tools and Analyses → Difficulty pulling large-scale data for batch GCS analysis using Pope/Mavrich 2017 scripts
| Link to this post | posted 09 Mar, 2022 01:04 | |
|---|---|
|
|
Hello, I am attempting to do a large-scale GCS comparison of all the phages in Cluster AZ (and plan to do this on other phages as well in the future). My former TA Andrew Kapinos wrote up a set of instructions to accompany the github code for the Pope & Mavrich 2017 batch GCS/MaxGCDGap scripts. I've been able to set up mySQL and other necessary components, and successfully pulled some data, but am stuck again. When I do the data pull from the Actino_Draft database using mySQL, I can't seem to pull the pham and function data that the GCS scripts require for the function.csv file, according to the instructions I have (I left comments in that file to highlight where I got stuck). Can anyone make a recommendation of what I might try next? Kudos to Nick for the help he's provided so far - thought I'd ask here as well so I'm not pestering him too much! (thanks Nick!) Best, Amanda |
Posted in: Bioinformatic Tools and Analyses → Difficulty pulling large-scale data for batch GCS analysis using Pope/Mavrich 2017 scripts
| Link to this post | posted 18 Feb, 2022 16:21 | |
|---|---|
|
|
Awesome! Thank you Christian. I will download the new one and email you with any questions I have. |
Posted in: Bioinformatic Tools and Analyses → PhamNexus on SEA-VM
| Link to this post | posted 17 Feb, 2022 23:31 | |
|---|---|
|
|
Hi all, I've followed the directions above in the SEA VM to try to install Phamnexus. It appears to have been cloned and installed, but when I click on PhamNexus, nothing happens… Not sure if it makes a difference, but I am using the version of the SEAVM downloaded here (https://seaphages.org/software/virtualmachine/) just a few weeks ago. I've attached a screenshot of the VM environment. Any advice is most welcome! - (As a second issue, I also tried the above trick to "Check for Updates" in the working version of PhamNexus we have installed on our computer lab computers, and the database did not update. Any recommendations for that would also be very helpful.) |
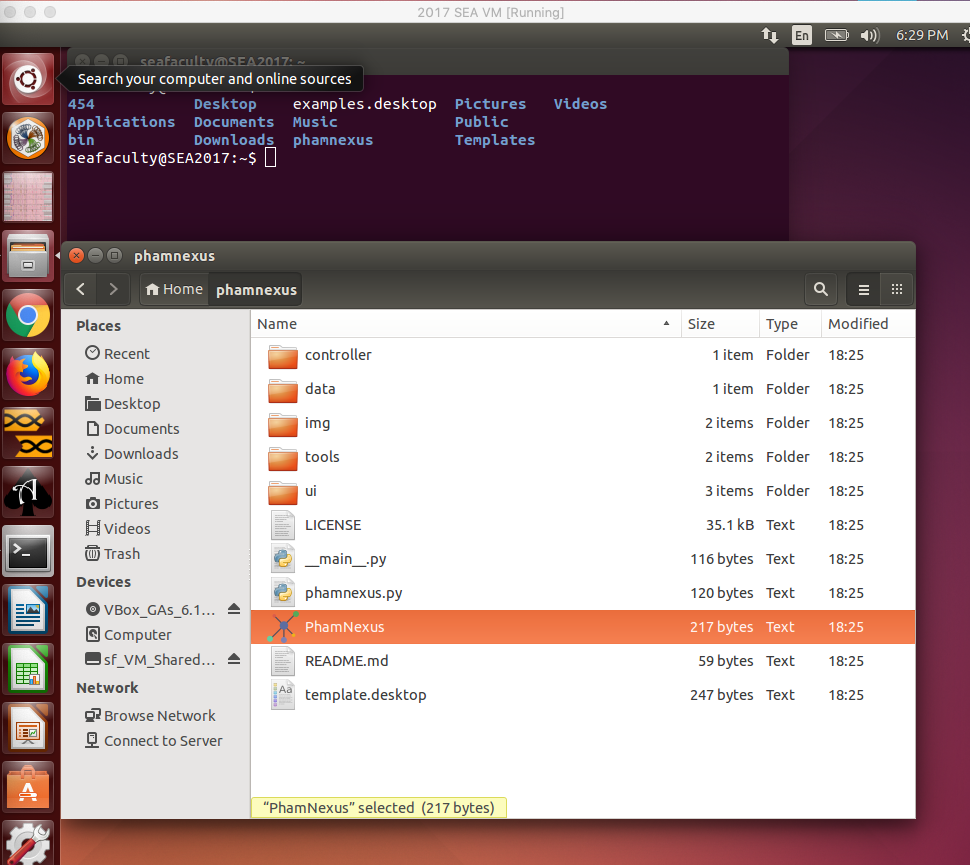 400Kb
400KbPosted in: Bioinformatic Tools and Analyses → PhamNexus on SEA-VM
| Link to this post | posted 11 Feb, 2022 02:37 | |
|---|---|
|
|
My guess is that Starterator just doesn't have the capability to say "there's a tie." It probably just chose the first of the two starts and designated that as the most-annotated one even though they are actually equal. |
Posted in: Starterator → How is the most annotated start determined when 2 starts have the same number of manual annotations in the same pham?
| Link to this post | posted 17 Nov, 2021 00:04 | |
|---|---|
|
|
Thank you for the question Arturo and the answer Debbie - we had the same question for our Cluster DE phage ObLaDi. Much appreciated! |
Posted in: Cluster EJ Annotation Tips → Potential minor capsid protein
| Link to this post | posted 12 Nov, 2021 16:18 | |
|---|---|
|
|
Hi all, thank you so much for this information! Like Kyle, I have several students interested in doing independent research projects, so I was considering having them pilot/optimize/test these hosts first to see if we are able to use them for PHAGES in the future. It would be great to meet and chat about good options for new hosts. Vic, I will send you an email. I'll CC you as well Kyle. Best, Amanda |
Posted in: Phage Discovery/Isolation → New hosts - human microbiome
| Link to this post | posted 11 Nov, 2021 22:51 | |
|---|---|
|
|
Thanks Kyle! Good to know. |
Posted in: Phage Discovery/Isolation → New hosts - human microbiome