Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by GregFrederick@letu.edu
| Link to this post | posted 18 Feb, 2016 18:30 | |
|---|---|

|
Lee HughesGregFrederick@letu.edu Thanks Lee. I had forgotten about that possibility. But does that mean that DaVinci (see image above) and all the others that do not have the programmed frameshift called are wrong and should be corrected? |
Posted in: DNA Master → LO: Designation Question
| Link to this post | posted 18 Feb, 2016 17:39 | |
|---|---|
|
|
Dan RussellGregFrederick@letu.eduDan Russell Thanks! |
Posted in: Gene or not a Gene → Cluster G genes spanning the COS site
| Link to this post | posted 17 Feb, 2016 22:34 | |
|---|---|
|
|
Dan Russell Dan: I do not have the names in front of me. But when I looked at it with students the other day it appeared that all the hits were in the NCBI Blasts or in 'draft' genomes in the PHAGESBD database. I concluded that those in the NCBI Blast hits were probably "auto-annotated" and deposited without QC (possibly by another group). We determined it was not likely a "real" gene. But it is an incredibly long ORF. So that call was not entirely 'comfortable'. |
Posted in: Gene or not a Gene → Cluster G genes spanning the COS site
| Link to this post | posted 17 Feb, 2016 19:38 | |
|---|---|
|
|
cdshaffer Great info! Thanks again! Greg |
Posted in: DNA Master → Genes Across COS sites???
| Link to this post | posted 16 Feb, 2016 23:22 | |
|---|---|
|
|
I have not looked yet. But are there any examples of genes spanning the COS site? We have about 1kb on the right end of one of our genomes that has no called features. Has anyone ever looked for genes that span the COS sites? Is there an easy way to do that? I'm just wondering if we should do so. Or if we might be missing genes there if we do not look. Thanks. |
Posted in: DNA Master → Genes Across COS sites???
| Link to this post | posted 16 Feb, 2016 23:21 | |
|---|---|
|
|
Another confusing one is two adjacent start codes. (I asked a friend on the QC team. But I guess I'll throw it out here too.) In some of the finished genomes in PhagesDB the first ATG is used. In some the second is called even if there is little or no overlap. The SD values seem almost identical. In most cases both starts seem to include the entire "coding potential". But only some of the finished genomes align 1:1. Others align 1:2 or 2:1. If everything else is the same, do you call the first start, the second, the one starterator prefers? Questions. Questions. Thanks for sharing your wisdom! |
Posted in: DNA Master → LO: Designation Question
| Link to this post | posted 16 Feb, 2016 22:31 | |
|---|---|
|
|
Here is an example of what is a "reasonable". All the of the phages we compared to Wunderphul are in the PhageDB phamerator DB. Look at feature 28. In Zaka and CLoudwang3 they apparently called the longest ORF. But based on guiding principles it does not seem "reasonable" because of the entire overlap with feature 27 in those two genomes. This one is easy to determine in our case because we have a STOP and this is the largest ORF in our case. But it obviously was not in Zaka and Cloudwang3. QUESTION: Do you want us to note those discrepancies so they can be edited? Are previous calls ever corrected/updated based on newer genomic information? There are multiple examples of numerous starts being used in the databasse for a lot of our genes so determining the best and most reasonable start call remains complex even if homology trumps everything. QUESTION: In these cases do we rely on the "longest reasonable ORF" strategy (unless it severely breaks one or more guiding principle like feature 28 above)? If one frequent call results in 20ishBP overlap and another frequent call results in 3-4bp overlap, does one choose the longer or the shorter? Obviously my "mental algorithm" is stuck in a loop and trying to add code that will help resolve the loop cycle. Thanks.  
|
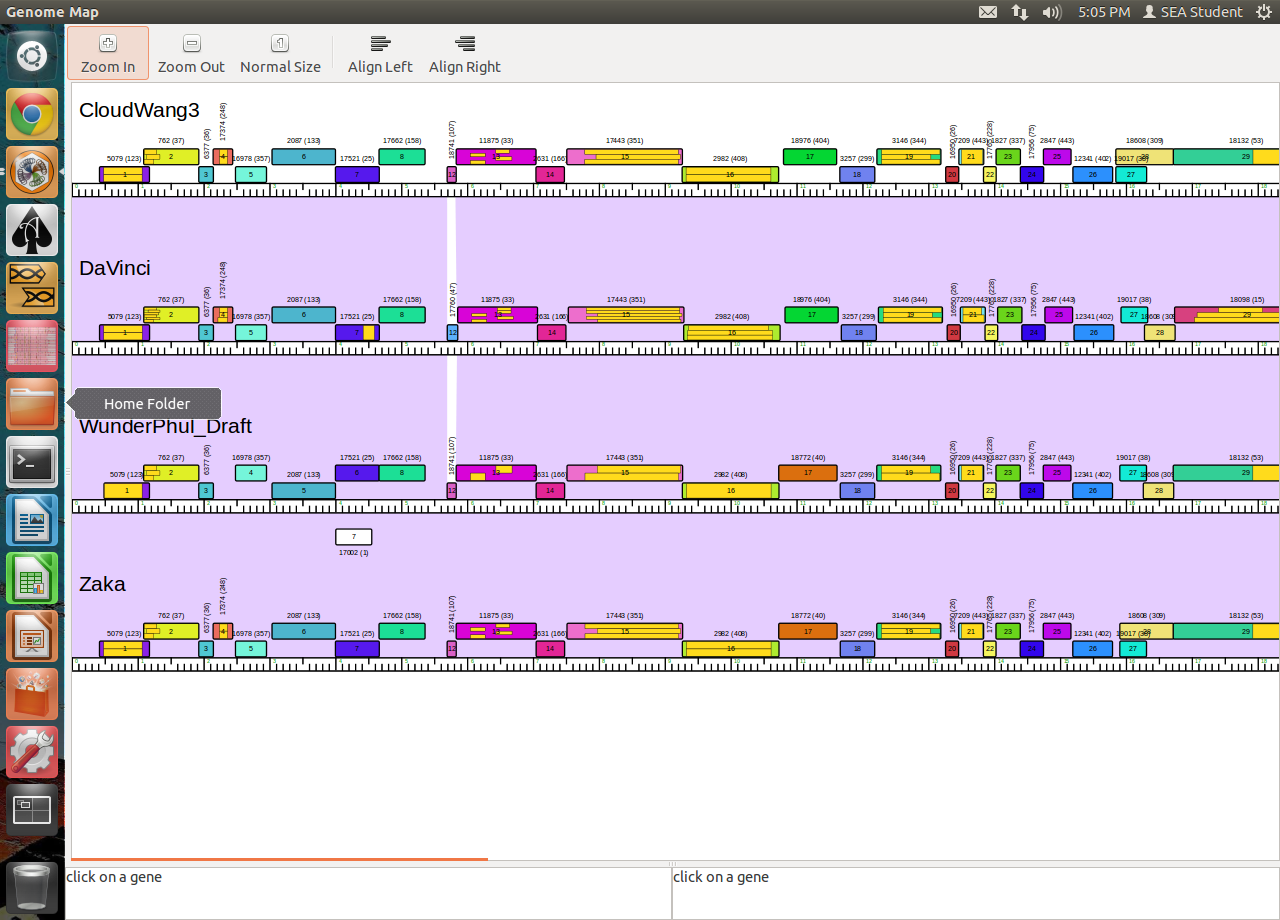 235Kb
235KbPosted in: DNA Master → LO: Designation Question
| Link to this post | posted 16 Feb, 2016 19:41 | |
|---|---|
|
|
Welkin Pope I hope that helps! Thanks Welkin! So what if a BlastP ends up showing both ORFs (or even more than two) in published, non-draft genomes? Do you suggest using the longest published/finished ORF? Even if it breaks one or more of the guiding principles? The most abundant BLASTP result or what? Deciding what is 'reasonable' can be complicated! |
Posted in: DNA Master → LO: Designation Question
| Link to this post | posted 16 Feb, 2016 16:09 | |
|---|---|
|
|
We want to make certain we are completing the information in DNA Master correctly. At our recent training we were given the two choices for the "LO:" description. LO: Longest Reasonable ORF -or- Not Longest Reasonable ORF (explain in notes below) I think we have some confusion over this language and a worksheet we 'borrowed' from another school. That worksheet actually has the student record the length of the longest open reading frame. Am I correct that the DNA Master "Notes" section does not require this information? When would one choose an ORF that is not the longest reasonable ORF? If there was a long overlap, that would make it unreasonable? (We are finding some 90+bp overlaps that have been used in PhageDB.) If the SD sequence was 3' of the ATG that would make it unreasonable? What else might cause one to select an ORF other than the "Longest Reasonable ORF"? Can you give me some scenarios that might lead one to select the "longest 'unreasonable' ORF" or a "shorter-than-longest reasonable ORF"? I hope these questions are reasonable! |
Posted in: DNA Master → LO: Designation Question
| Link to this post | posted 12 Feb, 2016 18:01 | |
|---|---|
|
|
cdshaffer Excellent. You are awesome! Just for information sake. It was crashing at feature 13 when we tried to run it. I don't know if that info will help with diagnosis or not… But now you have it! Thanks so much! Greg
|