Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
Debbie Jacobs-Sera posted in Validating Translational Frameshifts in DNA Master
storksle posted in Validating Translational Frameshifts in DNA Master
Debbie Jacobs-Sera posted in Validating Translational Frameshifts in DNA Master
Lee Hughes posted in Validating Translational Frameshifts in DNA Master
storksle posted in Validating Translational Frameshifts in DNA Master
All posts created by viknesh
| Link to this post | posted 15 Oct, 2025 17:03 | |
|---|---|

|
jayadasgupta The recommendation is to minimize the number of passages, as each passage increases the genetic variation in the population and possibly phenotypes important for infection. For a reliable culture, we recommend always starting a culture from a colonies off a freshly streaked plate. In a pinch, you can subculture from an existing culture, but I would do this regularly. |
Posted in: Phage Discovery/Isolation → Sub-culturing from PyCa Broth
| Link to this post | posted 06 Aug, 2025 16:27 | |
|---|---|
|
|
pia1@pitt.edu Hi Ping, The ordering information is available in the Ordering Guide, which you can access on the faculty info page. Here's a direct link: https://seaphages.org/faculty/information/#phagediscovery Best, Vic |
Posted in: Phage Discovery/Isolation → Ordering info for TEM work
| Link to this post | posted 21 May, 2025 14:09 | |
|---|---|
|
|
e.guevara Hi Elena, I suspect it is the culture. We've found that our plates were grainy when we grew the P1 culture (with Tween) for only 2 days. Letting the P1 culture incubate with shaking for 3 - 4 days alleviated the problem for us. We also grow our P2 for 3 days. Let us know if this helps with the grainy lawns you are seeing. Vic |
Posted in: Phage Discovery/Isolation → 2X smeg top agar
| Link to this post | posted 15 May, 2025 23:18 | |
|---|---|
|
|
jeremy Hi Jeremy, You are correct that UA is minimally radioactive, and is so because it is made from depleted uranium. For a 1% uranyl acetate solution (the stain we recommend), standard BSL1 PPE (gloves, lab coat, safety glasses) will suffice; this is true at many institutions and imaging facilities. The final say however has to come from your institution's safety office. Vic |
Posted in: Phage Discovery/Isolation → Phage Staining - Uranyl Acetate
| Link to this post | posted 26 Feb, 2025 21:09 | |
|---|---|
|
|
JustinA Hi Justin, I think Adam Rudner at Ottawa has generated lysogens for a couple of AY phages – you might want to reach out to him, or better, invite him to respond here. I'd also like to suggest generating lysogens using the plate-seeded method, now included in the Phage Discovery Guide and listed as part of the methodology for our program-wide effort to raise lysogens – see the lysogen datacard template on QUBES: https://qubeshub.org/community/groups/hhmi_sea/research_data_collections Best, Vic |
| Link to this post | posted 04 Feb, 2025 21:46 | |
|---|---|
|
|
kcornely Hi Kathleen, The Fisher quote on seaphages.org includes albumin. You can access the quote here, under Phage Discvery component: https://seaphages.org/faculty/information/ Vic |
Posted in: Mycobacterium → Source of albumin for AD
| Link to this post | posted 28 Jan, 2025 02:59 | |
|---|---|
|
|
Thanks to a collective effort across SEA, we have found that the Fisher brand of peptone is the culprit for the precipitation that many are observing in their PYCa media. In fact, there is another brand of peptone by Fisher called "peptone for science education" which make PYCa media crash immediately after it is sterilized in an autoclave. The SEA Team (thanks, Cole Jirsa!) has now tested multiple brands of peptone for precipitation, and we recommend any of the following 4 brands of peptone, which we've listed from most expensive to least expensive : Bacto Peptone (Difco) , MP Biomedical Casein Peptone, Neogen Peptone, and HIMEDIA Peptone. We also note that there are no observable differences in the efficiency of plaquing or plaque morphology for at least three different phages when plated on media made using any of these 4 brands of peptone. We'll try to include these brands of peptone in the Fisher quote as soon as possible. If you switch to any of these brands of peptone, please share with us any notable observations! |
| Link to this post | posted 22 Jan, 2025 19:32 | |
|---|---|
|
|
stpage Hi Shallee, It looks to me that you are indeed seeing individual plaques form for the one dilution after the last spot that resulted in a full clearing. However, it looks like your lawn is not particularly even. As a result, it is hard to see those plaques. Because those plaques are forming within a defined spot on the plate, it is not too hard to see those plaque when we compare that defined area to the rest of the plate. However, I imagine that we wouldnt be able to see those plaques if they were distributed across the entire plate, as they would be in a plaque assay. So the first thing we need is a nice lawn. I suspect the lawn is uneven due to the top agar. I would suggest melting new/fresh top agar and preparing a top agar lawn with freshly prepared bacteria too. I think you'll then be able to see the plaques. Hope this is helpful. Vic |
| Link to this post | posted 10 Dec, 2024 19:21 | |
|---|---|
|
|
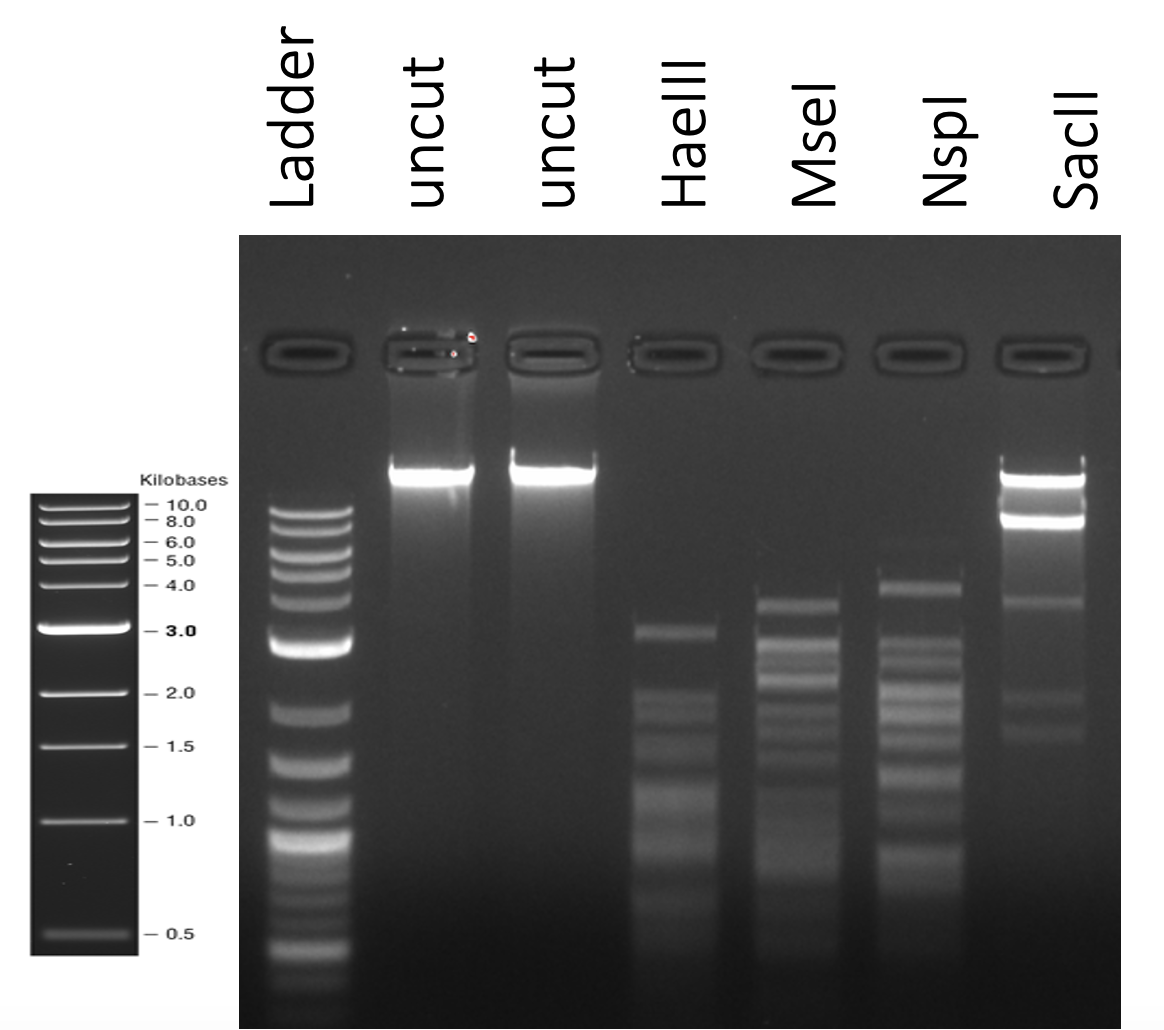
nic.vegaThanks for sharing this – very nice gel. If your student is willing to do it one more time with a third setup: Regular 1 hr; Regular 1 min, MWV 1 min, that'd be helpful. Even in our regular setup (no microwave),I think it is likely that the majority of DNA is restricted within the first minute, which might potentially give us the same results as the MWV reaction. Thanks, Nic. Vic |
Posted in: Phage Discovery/Isolation → did you know you can do restriction digests in the microwave?
| Link to this post | posted 05 Dec, 2024 22:02 | |
|---|---|
|
|
Hi Nic, It might be useful to gather more side-by-side data for digests done using our standard method vs the microwave method. It might be quicker to use the microwave, but it’s unclear to me if the quality of the data is where we need it to be (I appreciate that the gel might just be a litle smeary). At the moment, we are beginning to look for phage DNA modifications, which can be gleaned by looking for inconsistencies in the expected digest pattern (i.e. a virtual digest based on the DNA sequence) vs pattern observed when the actual digest is run on a gel. If it is tricky to get good digests and gels using the microwave method, then that’ll impact the utility of the data for that purpose. The TrixiePhatell gel from a regular digest can be viewed here. We’re always excited to hear of new ways of doing our collective research. As we learn of new strategies though, the SEA team usually invests time to understand the implications of the new technique for our collective work before we incorporate anything into our regular workflow. This is so we can continue to leverage the large datasets that are generated by faculty and students from across the program to glean new insights. If some of that data is generated differently, it can impact our interpretation. So if you are willing to gather some data to be reviewed, perhaps over the next semester, please do share. Best, Vic |
{kind=link}