Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by uOttawaPHAGE
| Link to this post | posted 02 Feb, 2024 14:24 | |
|---|---|
|
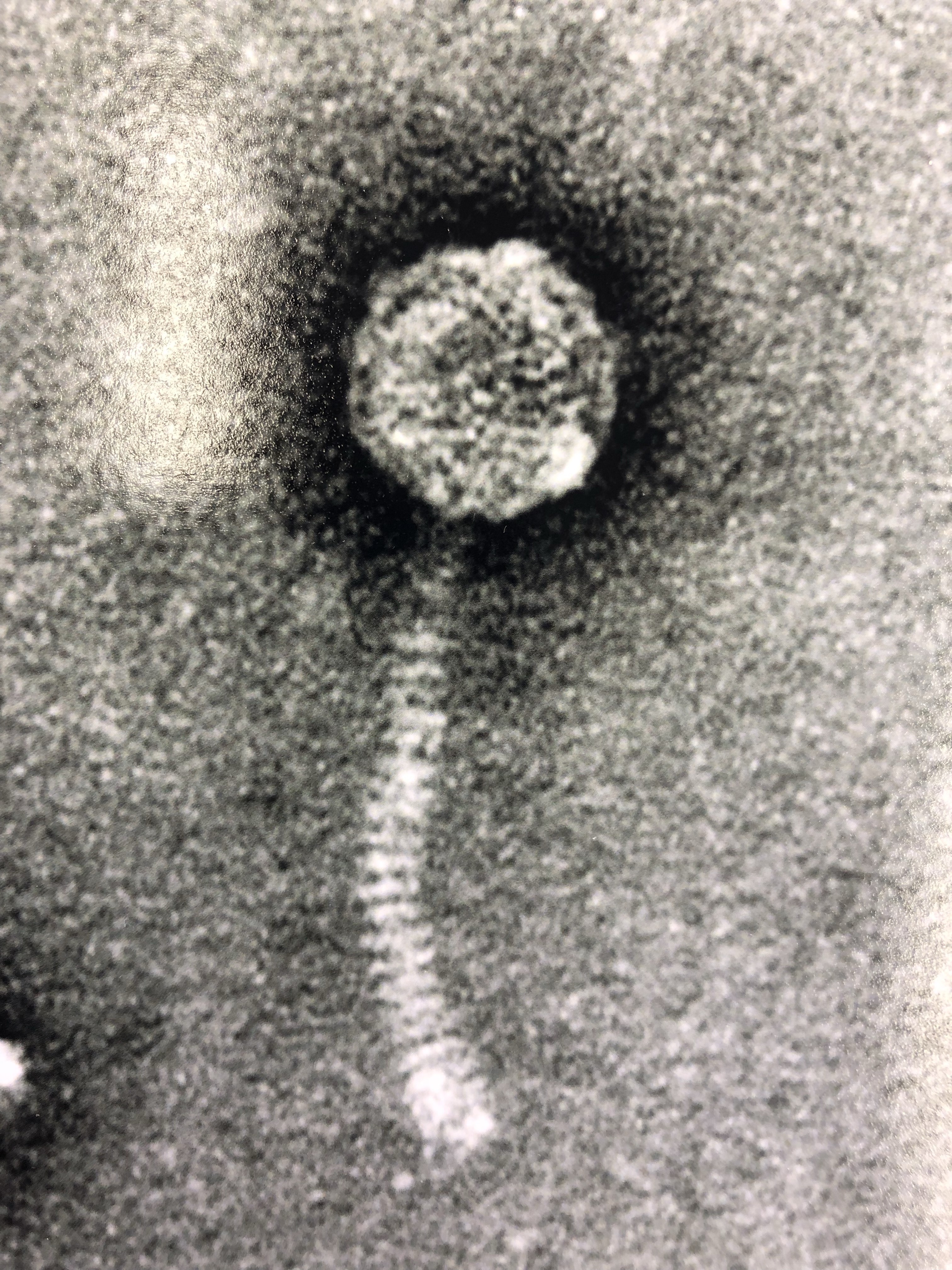
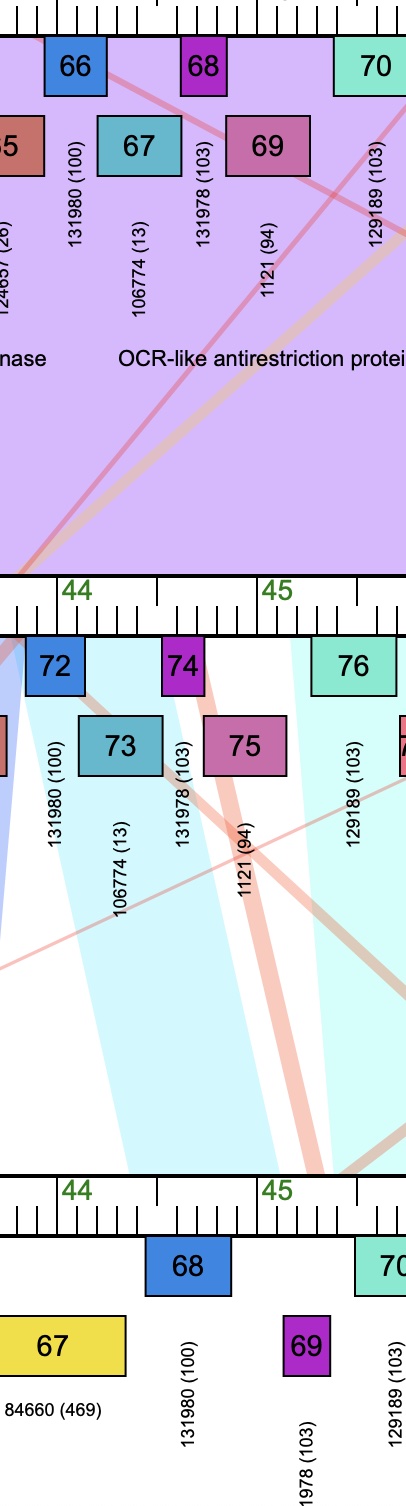
hi Debbie. To not lose coding potential in gp72, the student selected the start site at 44271 (also has better scores/spacer) and this gives a 158bp overlap. It sounds like you are suggesting we use 44145 to minimize overlap, and not worry about cutting off coding potential? Part of why I posted this is because there is a clear difference in the way the other related genomes were annotated - one QCer added a gene, and I'm guessing the other decided the overlap was too big an issue. I didn't get into the functional calls, which are obviously important, but I first wanted to get an opinion on the 158bp overlap. Phamerator map screenshot attached. Diane on top, Superstar_draft middle, Issmi bottom. Adam |
 125Kb
125KbPosted in: Annotation → Gap or overlap in Superstar (BD2)
| Link to this post | posted 02 Feb, 2024 01:18 | |
|---|---|
|
|
We are annotating phage Superstar (BD2) and aren't sure if we should call a large overlap (~150bp) between gp73 and gp72 (~44,250), or a large gap (~300bp) by deleting gp73. Some phages (Ismmi) have deleted the gene, others (Diane) have kept it in. Good coding potential in gp73 (and in the two comparators) that ends abruptly where gp72 begins, but no STOP for ~150bp. My bias (unfounded? Maybe based on my R2I session with Sally Molloy?) is to fill the gap, trust the coding potential, and accept the overlap. Or should we delete gp73? The QCers have had different opinions (though the overlaps do seem to vary somewhat - perhaps more evidence for the idea below). One solution is if there is a programmed slip, and this is "called" (the black triangle) in the Genemark files of Diane and Issmi (actually Diane calls slips between the gp74-gp73-gp72!). My student found a potential slippery sequence in the correct location in Superstar, and we may test it in E. coli, but I know without evidence we shouldn't call this. Any advice/opinions? Adam |
 143Kb
143KbPosted in: Annotation → Gap or overlap in Superstar (BD2)
| Link to this post | posted 03 Oct, 2023 15:59 | |
|---|---|
|
|
Hi Bernadine. Based on an email exchange with Kyle (posted below), he thinks his issue was contamination with Geobacillus stearothermophilus, a thermophile that grows at 55-60 degrees and forms spores, so something that could be hard to get rid of. I also learned that these spores are used to test autoclaves and it is ubiquitous in the environment. My current thinking is our autoclave may not be killing every last spore, but we only see this in our top agar. We've increased the cycle time to 55' until we can have the autoclave checked. We wouldn't see problems in our plates/liquid (unless we put them at 55 - we are trying this) because the bacteria won't grow at 30-37. Not sure if you run the spore tests for your autoclave, but you might want to. Kyle is trying to avoid an extended time at 55, which may work as well, but we can't really make that work for our course. Good luck! Adam Kyle's email to me: Happy to talk this out sometime if it’s helpful. I went to reply on the Forum but I need to reset my password and just decided to do this instead. Re-reading the forum entries and both remembering things from our first bout and also probably forgetting some things, I can say that I feel pretty sure of what was the original cause of our problem. We isolated bacteria from the top agar. We sequenced it. It was Geobacillus stearothermophilus. Naturally, as a thermophile this makes sense. But where did it come from? I believe for us the source was our still. We still have an old distilled water still in our prep lab. We have always used it to make media with. For making regular old media to use at 37C, the presence of Geobacillus would never be noticed. But our phage work is the ONLY work with a hold step at 55C, which is just getting a Geobacillus going…. Given that we have sequencing data to show it is definitely this species, I had our lab manager replace the outflow hose from the still and the problem seemed to go away. Now I give this all to you as preface because, lo and behold!, it is BACK, RIGHT NOW. My TAs this semester were IN the course in SP21 when we had all the problems. And they are very very careful. We’ve done the usual stuff, replacing everything, using water from a different system we have upstairs, autoclaving the hell out of everything, before using containers and then again with media in them. We’ve put the TA in disposable sterile 50 ml conicals. We get contamination everywhere. It seems for us like ANY hold step at 55C is just asking for trouble. Vic suggested at first it could just be calcium precipitation and not contamination, but this looks really biological based on how it grows in the bottles. I am at the moment convinced the only thing we can do is basically never hold the TA at 55C except for the shortest period of time possible. So my plan for Monday is this: We made a couple large glass bottles of top agar. All the usual safeguards, autoclaved the hell out of it, and now we have let the TA bottles cool, and have placed them in the fridge to be solid, happy, and definitely not growing thermophiles, over the weekend. On Monday, we will boil the top agar to liquefy it, then let it cool, aliquot into sterile conicals, and place in the 55C bead baths. When the students are done with their Monday TA we will have them dispose of the TA. We will then rinse and repeat for Weds. This seems like vast overkill, but unless we absolutely minimize our TA time in 55C it seems like a continuous problem right now. Sounds crazy, doesn’t it? We also don’t see contamination in our plates OR in broth. Only the TA. |
| Link to this post | posted 29 Sep, 2023 03:47 | |
|---|---|
|
|
Hi Kyle. We think we are having an issue similar to what you describe above. We have just tried new agar and it seems to have helped but not completely. Given the contaminant survives the autoclave, it could be in some of the bottles that are filled with top agar, so I suppose it could persist. I’m hopeful this will fix the problem. Two questions: 1. In your thread you mention you were also having issues with phage propagation, which it sounded like you linked to the top agar issue. Did the propagation issue resolve when the top agar issue resolved? I’m assuming yes, but you didn't say it above. We are seeing propagation issues, but only for a subset of phages. The phages were found on new hosts for us, A. glob 2979 and A. sulf, and we aren't sure if this issue could be more common for these hosts, or if it is linked to the top agar. 2. We aren't seeing contamination in our plates, which is confusing to me. We wonder if this could be because of the cycloheximide. Did you ever consider this? This would suggest a fungal contaminant. Adam |
| Link to this post | posted 14 Jul, 2023 06:44 | |
|---|---|
|
|
mdgainey hi Maria. I have inquired at my institution and uranyl acetate seems to exist in a nether state between radioactive and chemical waste, and neither of the individuals responsible for these two wastes want to claim it. Hopefully it will be more straightforward at your institution. |
Posted in: General Message Board → Electron Microscopy
| Link to this post | posted 11 Apr, 2023 19:17 | |
|---|---|
|
|
Hi Michèle. I quickly looked at the HHpred results for these two genes. IF you look down your list their are hits with HTH domains, and many of your hits are either sigma factors or related to sigma factors, which have HTH domains. Upon closer inspection the matches all appear to be minimal domains that have three (in one or two cases only two) alpha helical domains with short non-helical segments. I believe they both meet the threshold of having a HTH domain. I still struggle with what this function should be. I think "HTH domain" or ""HTH domain containing protein" would be best, but I don't thinks these are options on the approved function list. Many of the hits are to transcription factors that have HTH domains, so they could be "HTH DNA binding domain protein" but it does seem like there isn't sufficient data to call these "DNA binding proteins." Debbie will need to weigh in on this. It would seem like we need a new more minimal function for these types of proteins. Adam |
Posted in: Annotation → helix-turn-helix binding domain or protein?
| Link to this post | posted 28 Mar, 2023 20:04 | |
|---|---|
|
|
thanks Debbie. We will leave it NKF and I'll suggest the students see what they can learn from published work or if any of the foldseek matches are cryoEMs from a podo. I also suggested they see if some of the other T4 proteins (gp11?) is present in the genome. I was mainly struck by the alphafold/HHpred similarity, but it sounds like there is currently insufficient data to give it a specific call. Have you gotten similar questions from all annotators of this pham? Adam |
Posted in: Annotation → Kikiko_42
| Link to this post | posted 24 Mar, 2023 20:18 | |
|---|---|
|
|
Kikiko_42 (46820-49244) is in a pham that has been called NKF in all other annotations, but it has very strong HHpred hits to gp12 in T7 (and other phages) that is part of the DNA ejection machinery. On Alphafold it also has very high probability to have a similar structure to gp12. I'm trying to understand why it has no approved function despite - is it because unlike T7, which has a contractile tail, this gene is found in podoviridaes? Without experimental data do we leave it as an NKF? Given that this pham has many members, I assume you have answered this question before…. Adam |
Posted in: Annotation → Kikiko_42
| Link to this post | posted 03 Feb, 2023 19:13 | |
|---|---|
|
|
I have a similar question about Polkaroo, a P1 cluster phage. Polkaroo_12 (7912-8325) (current pham# 68611) is annotated as NKF for virtually every pham member, but has high HHpred hits to a putative tail component and a minor capsid protein. Similar hits to HK97_10, but also other proteins. See: https://www.ebi.ac.uk/interpro/entry/InterPro/IPR021080/ Why are so many of the hits calling it a minor capsid protein? |
Posted in: Cluster EJ Annotation Tips → Potential minor capsid protein
| Link to this post | posted 02 Nov, 2022 20:30 | |
|---|---|
|
|
Hi Dan. We're finally about to do our inaugural MiSeq run with ~20 libraries. A few final questions, if you have time: 1. We were going to use the Illumina Protocol A for denaturing/loading - I'm assuming using Bioanalyzer results constitutes "standard normalization"? 2. Do you denature 5uL of 4nM pooled libraries? 3. And to clarify, with 20 libraries each at 4nM, that would only be 0.25uL of each library. 4. IN a response above you mentioned your libraries are often not much above 4nM. Ours are higher, so I'm wondering if we did our library prep differently. We used the NEBNext Ultra II FS kit. How much input genome to you use? 5. Do you add 1% PhiX as a control? This seems standard. 6. Any other subtleties I should know about? thanks again for your help with these questions. Adam |