Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
cdshaffer posted in Whole phage starterator reports
Debbie Jacobs-Sera posted in frameshifts
uOttawaPHAGE posted in frameshifts
Dan Russell posted in Congrats to Steve Caruso and Beth Wilkes — 2025 ASM Outstanding Instructor Award, Honorable Mention
ACMPhageHunters posted in Clarification Question About HNH Endonuclease Function Determination in view of hits to the Ref Sequences
All posts created by stpage
| Link to this post | posted 11 Aug, 2025 12:55 | |
|---|---|

|
Hi, has anyone done kinetics assays on the phage replication using chloroform? We got some discrepant results….. |
Posted in: Phage Biology → chloroform
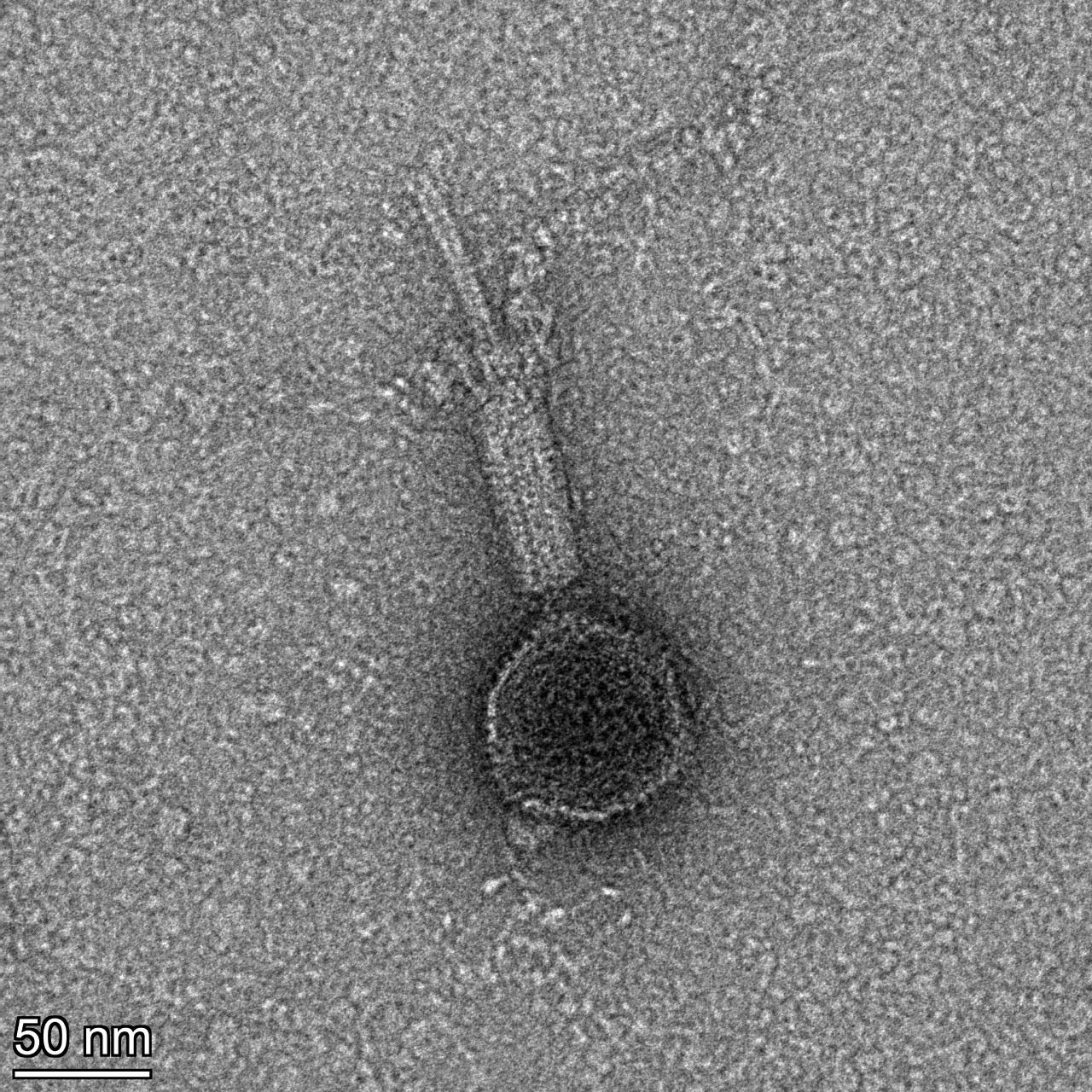
| Link to this post | posted 22 Jul, 2025 13:12 | |
|---|---|
|
|
Contracted tail |
 533Kb
533KbPosted in: Phage Biology → tailproteinsunwinding?
| Link to this post | posted 22 Jul, 2025 13:11 | |
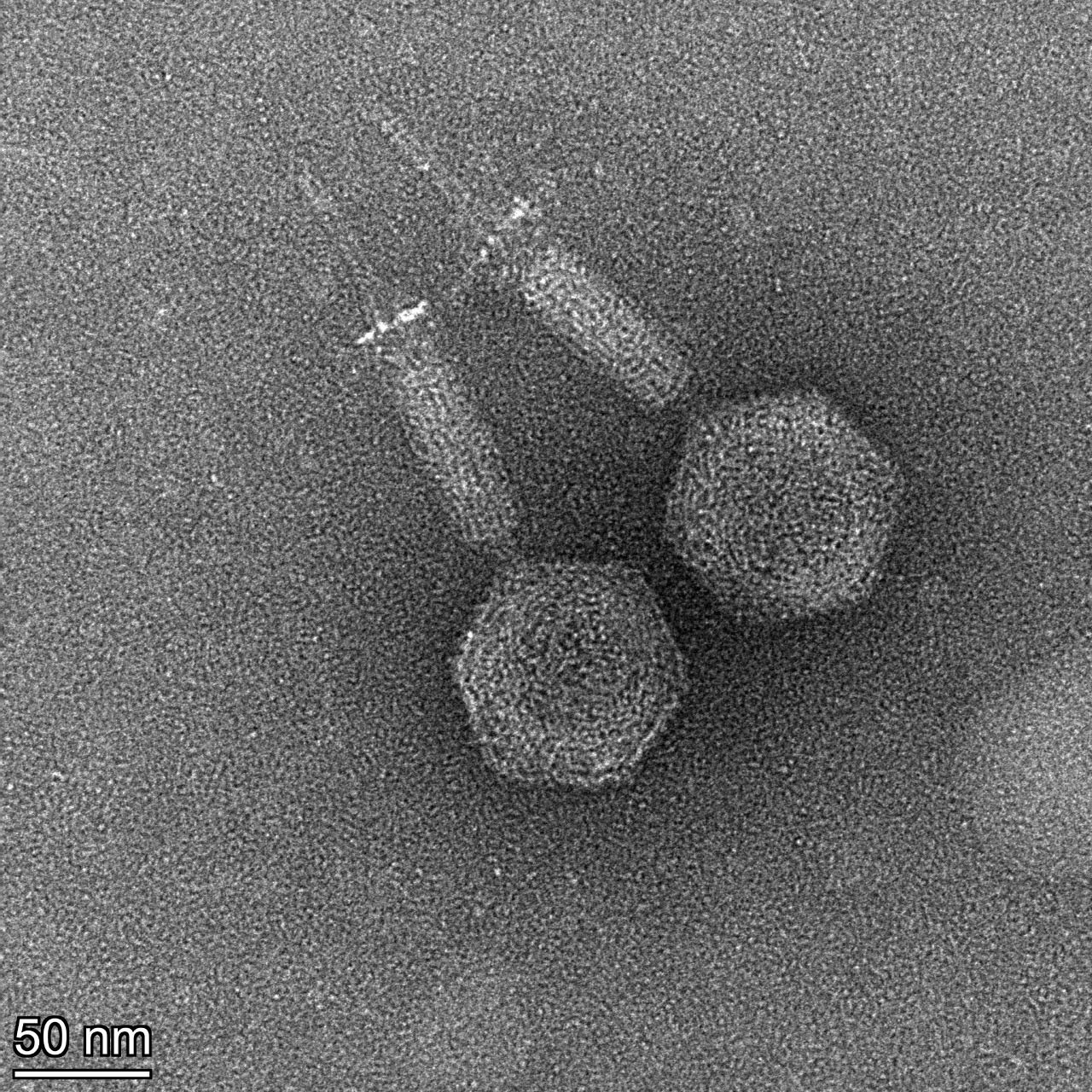
|---|---|
|
|
Hi, I have some fun TEM pictures, one typical, one with the tail contracted and one with the tail protein (like the gp18 of T4) that is unwinding off of the center stalk? (or maybe contracted with a long fiber of schmutz behind it) |
 501Kb
501KbPosted in: Phage Biology → tailproteinsunwinding?
| Link to this post | posted 12 Jun, 2025 18:15 | |
|---|---|
|
|
I think I would say in general that the pleskstrin-homology domains closely share a structure, but do not closely share sequence similarity *nor function*. So, not super useful annotation anyway. See: Mark A. Lemmon, Chapter 136 - Pleckstrin Homology (PH) Domains, Editor(s): Ralph A. Bradshaw, Edward A. Dennis, Handbook of Cell Signaling (Second Edition), Academic Press, 2010, Pages 1093-1101, ISBN 9780123741455, https://doi.org/10.1016/B978-0-12-374145-5.00136-4. (https://www.sciencedirect.com/science/article/pii/B9780123741455001364) |
Posted in: Request a new function on the SEA-PHAGES official list → Pleckstrin homology-like domain protein
| Link to this post | posted 20 May, 2025 18:36 | |
|---|---|
|
|
I ran alpha fold and HHPRED and am not super-convinced. It's only a beta-alpha-beta so pretty small compared to the other structures. But, the closest match in both HHPRED and alphafold is to a bacterial PH domain. So, certainly PH-domain-like. See attachment. |
 1.45Mb
1.45MbPosted in: Request a new function on the SEA-PHAGES official list → Pleckstrin homology-like domain protein
| Link to this post | posted 04 Apr, 2025 08:51 | |
|---|---|
|
|
mthomas Can you post the HHPRED coverage and percent identity? What do you get when you do an amino acid sequence alignment against the SAM lyase from T3? |
Posted in: Cluster ED Annotation Tips → SAM lyase ?
| Link to this post | posted 03 Apr, 2025 16:38 | |
|---|---|
|
|
Hi, here is a student tutorial I wrote for the Genome Education partnership. If you have any questions or suggestions, drop me an email! |
Posted in: Annotation → Alphafold tutorial
| Link to this post | posted 03 Mar, 2025 19:41 | |
|---|---|
|
|
Hi is there a standard protocol for reviving phage from -80 stocks? Well, I assume there is.  What I mean is, what do y'all recommend? What I mean is, what do y'all recommend?
|
Posted in: Phage Discovery/Isolation → Reviving
| Link to this post | posted 22 Jan, 2025 20:42 | |
|---|---|
|
|
Our A.glob lawns are often like that but we have been using top agar from early in last semester. We'll give that a try. Thanks, Vic! |
| Link to this post | posted 22 Jan, 2025 19:10 | |
|---|---|
|
|
So, we have had a lot of trouble amplifying, with great spot tests and poor plaque assays (a lot of killing at high concentration but no nice plaques at any concentration). I suspect lysis from without. So, after checking to make sure that we are plating from a single colony of the correct host, what would be the next step? Some spot test plate pictures attached. |
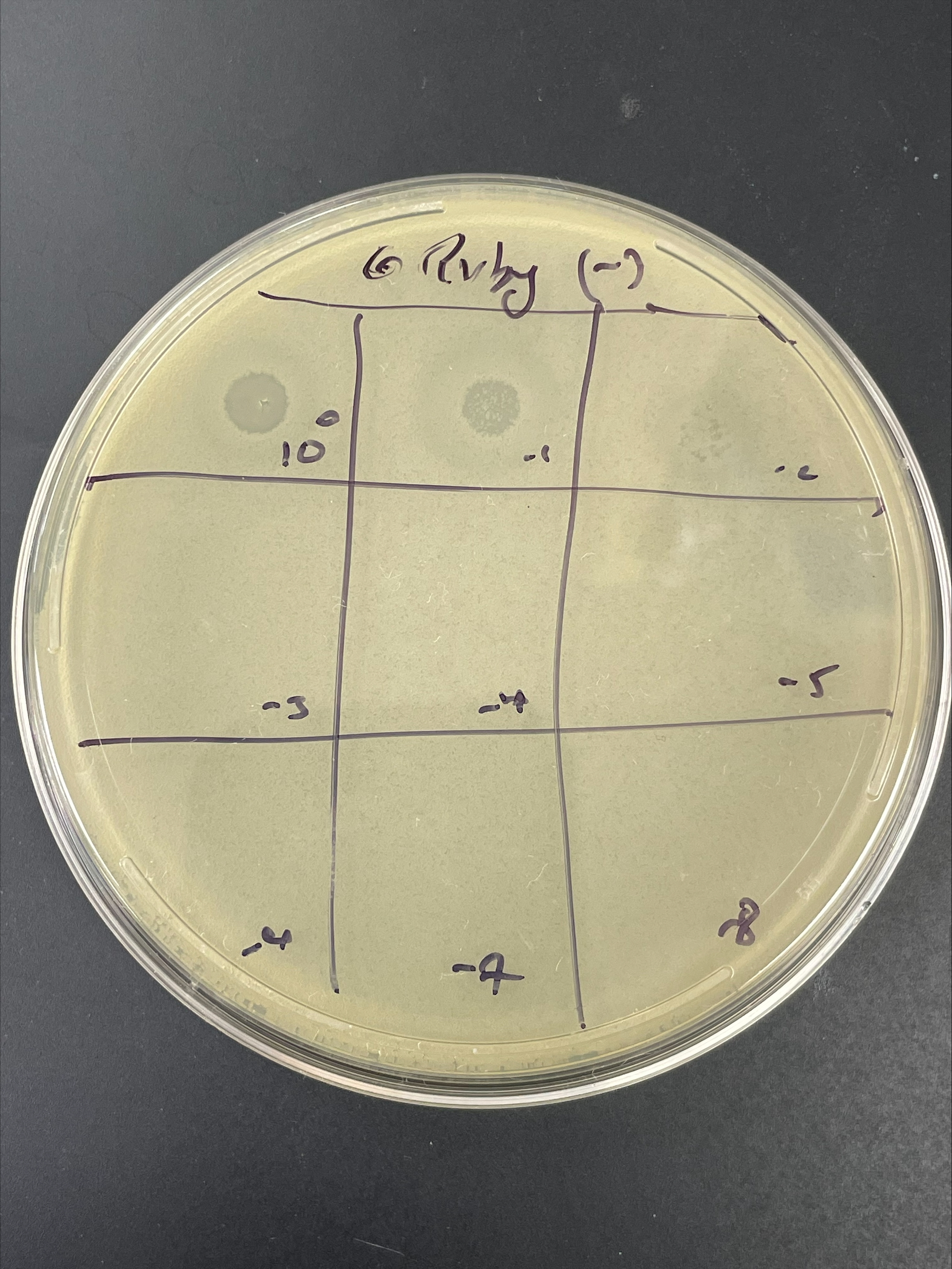 2.41Mb
2.41Mb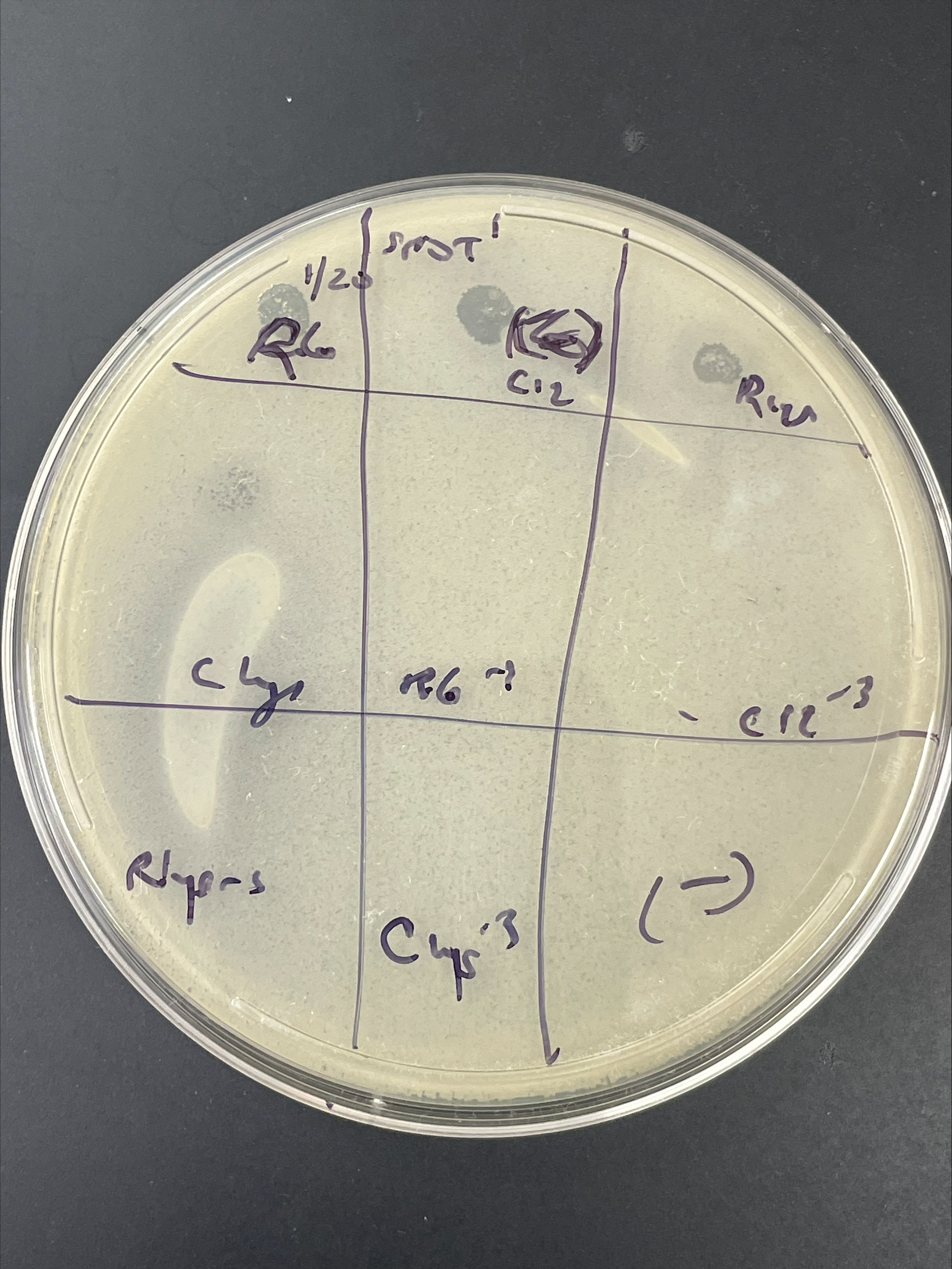 2.44Mb
2.44Mb