Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by MSMC
| Link to this post | posted 07 Sep, 2018 14:34 | |
|---|---|

|
I had discussions with multiple people at the symposium and faculty retreat about issues with extracting DNA from M. foliorum phages. It seemed that many people were running into an issue with increased smearing of the phage DNA when the digests were run on a gel. Has anyone tried any modifications to the protocol? We did one pilot where we omitted DNase, it did not have a large effect (only tried once though). I was thinking of increasing the time of guanadinium thiocyanate exposure a bit. Any thoughts? |
| Link to this post | posted 22 Jun, 2018 11:08 | |
|---|---|
|
|
That makes sense, thanks Debbie! |
| Link to this post | posted 21 Jun, 2018 20:18 | |
|---|---|
|
|
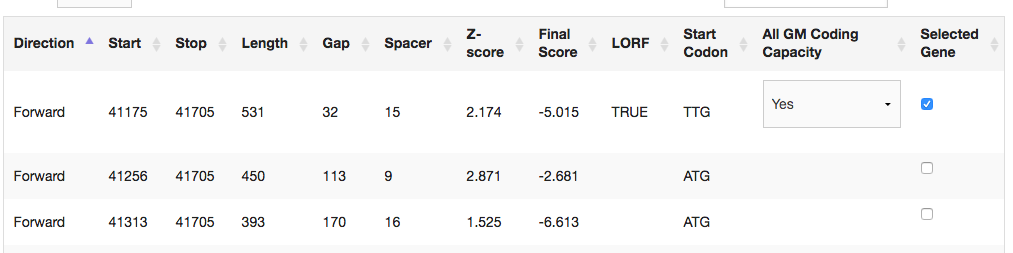
In looking at the coding potential for potential start sites, I was under the assumption that GeneMark identified all potential start sites (not the coding potential, but the ATG, TTG and GTG start sites). For this gene, the start with the longer ORF at 41175, which is annotated more often, does not show up on the GeneMark output. Is there a reason GeneMark would omit a potential start site? Maybe it does this and I haven't noticed in the past. |
 46Kb
46Kb 39Kb
39Kb| Link to this post | posted 13 Jun, 2018 19:33 | |
|---|---|
|
|
Thanks so much Chris, that helps me understand it much better. |
Posted in: PECAAN → Issues with BLAST results in PECAAN
| Link to this post | posted 12 Jun, 2018 19:08 | |
|---|---|
|
|
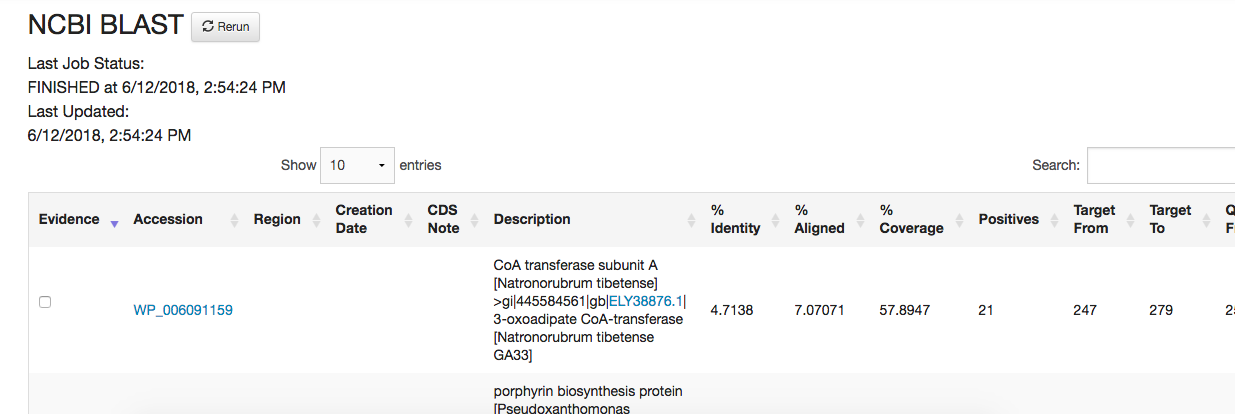
After the symposium and faculty meeting, I'm definitely sold on using PECAAN for my annotations. I'm currently annotating a phage and I'm having a bit of trouble with the way the BLAST results work. 1) Most of my NCBI BLAST results don't have any phage alignments, though they do show up in the DNA Master BLAST (see attachment). Do I need to change settings or something to get these results to mirror those that I get in DNA Master? 2) PECAAN is exporting BLAST results whose boxes are not checked. My understanding is that only the check information would be exported to be used in parsing the DNA Master file. Any help is greatly appreciated! Cheers, Evan |
 85Kb
85Kb 17Kb
17KbPosted in: PECAAN → Issues with BLAST results in PECAAN
| Link to this post | posted 08 May, 2018 19:35 | |
|---|---|
|
|
Kristen Butela Thanks Kristen, it definitely seems to be a memory issue, as it's now giving me the "not enough memory" error. It's only this phage, and it is the only file open. In the past I only used a virtual machine and the PC version of DNA Master for this genome, but I don't have the virtual machine on my computer anymore. I just sent you the file, any help is greatly appreciated!  Evan |
Posted in: DNA Master → Running DNA Master on a Mac using Wine
| Link to this post | posted 08 May, 2018 18:33 | |
|---|---|
|
|
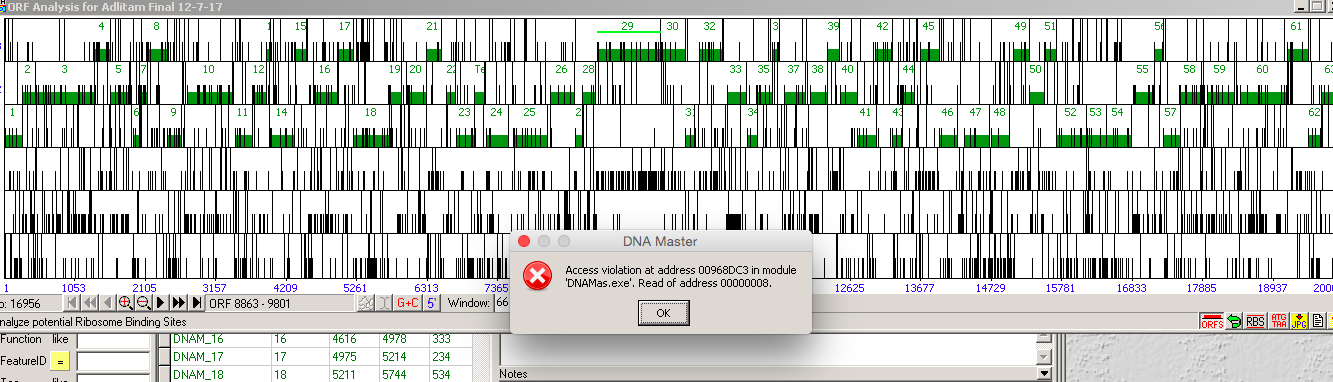
Second error message. |
 64Kb
64KbPosted in: DNA Master → Running DNA Master on a Mac using Wine
| Link to this post | posted 08 May, 2018 18:32 | |
|---|---|
|
|
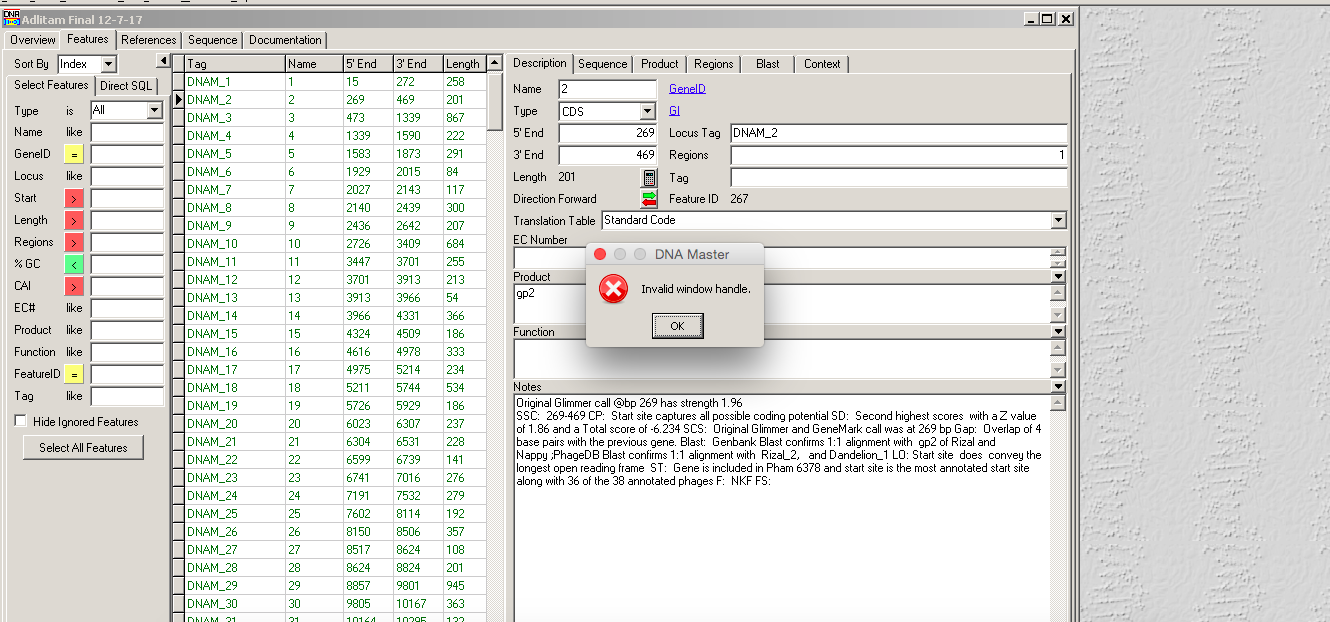
I'm having an issue working with one of my phages on WINE. When I open the file and click frames, it gives me an error that says "Invalid window handle" (pic attached). I can bypass this by clicking ok and then "ORFs", and it shows me the predicted ORFs. However, when I highlight one and click on the RBS, if gives me an error that says "Access violation at 00968DC3…" (pic attached on next post). This error only seems to come up with this C cluster phage, I'm wondering if it has to do with the size of the genome (the original error DNA Master gave me was "not enough memory" ). Has anyone else seen this? Thanks! Evan |
 171Kb
171KbPosted in: DNA Master → Running DNA Master on a Mac using Wine
| Link to this post | posted 05 Apr, 2018 14:29 | |
|---|---|
|
|
Debbie Jacobs-Sera I'm going to have my students look a little closer at this. If there is no programmed frameshift, is there a reason they two different genes would have the same pham #? |
| Link to this post | posted 20 Feb, 2018 11:28 | |
|---|---|
|
|
Bringing back an old thread here: We are annotating a phage from the host Microbacterium foliorum. When we get to the tail assembly chaperone genes (15 and 16 in Schnapsidee), we see what could be a programmed frame shift (two genes, both coding for tail assembly chaperone, of the same Pham, with overlapping open reading frames). However, in Phamerator, all of the EA1 subcluster Microbacteriophages do not indicate a programmed frame shift. I have not had an opportunity to look at the specific sequences yet, but have programmed frame shifts been observed in phages from hosts other than Mycobacterium? If this is not a programmed frame shift, is it strange for the two genes to have the same Pham number? |