Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by ClaireRinehart
| Link to this post | posted 10 Apr, 2019 16:55 | |
|---|---|

|
JoAnn, We just processed Chotabhai from PECAAN into DNA Master and then into the submission pipeline without any problems of getting the Hypothetical Protein tag to populate. Usually in these situations, we have found that the software that you are using to copy the file and paste it into the DNA Master documentation is inserting a character that is not compatible with the DNA Master parsing. Would you please copy the PECAAN "Export CDS Function" file and paste it into a new file, save it and then send it to us so that we can compare to our processed file. email to claire.rinehart@wku.edu. Please also indicate what software package you are using to view the PECAAN "Export CDS Function" file and to copy from. Thanks, Claire |
Posted in: PECAAN → New Features in PECAAN
| Link to this post | posted 11 Mar, 2019 14:46 | |
|---|---|
|
|
I am hoping you have a resolution for this. Debbie, We have added some modifications to PECAAN that will allow you to easily resolve this problem of having duplicate phage names. In fact, I recently made seven copies of a phage, Lilizi, that we had annotated previously but had not submitted, so that I could give students a phage to practice on that had the Pham and Starterator information attached, unlike Etude. For your Anthony phage, I would create a phage entry named Anthony_Myco and attach the files as usual. Information after the _ is usually ignored and the pham searches will be done with just the Anthony part of the name. If you are doing training and want to change the first part of the name to something that people won’t recognize as being the same phage you could enter something like TestSet_1 and then go into the Admin->Phages menu and then search for your phage TestSet_1 and press the Edit button at the end of the line. In the Edit window there is now an option to assign the Phamerator Phage Match that should be mapped onto the phage. This will also populate the Starterator field in PECAAN. Please be careful with this option and make sure that you are mapping the Phamerated phage name to a PECAAN copy with the same sequence. Give it a try and give us feedback. Thanks, Claire |
Posted in: PECAAN → New Features in PECAAN
| Link to this post | posted 11 Mar, 2019 14:37 | |
|---|---|
|
|
Steve, The phagesdb Function table that we have added provides a summary of the top 100 hits that we draw from Phagesdb. Those that show functions other than "Unknown Function" are when grouped by function pham and cluster to give you a summary of the evidence for each individual combination of those three elements. The Phagesdb Function Frequency is simply the number of hits for each line divided by the total number of functional hits in the top 100. As for differences between what you see in Phagesdb and what you see in the NCBI BLASTp, we can often see phages in Phagesdb that we don't see in the NCBI output because NCBI only shows one representative hit of a group of identical proteins. You can often see these by clicking on the NCBI BLAST Accession link for a phage and then clicking on the "Identical Proteins" button on the upper left of the gene window. We try to show members from this list in our Description field but are limited to the number that we have chosen to display, so, if you see a very long list, click on the Accession link and check out the "Identical Proteins" because we have probably not listed all of them. Differences between PECAAN hits and those obtained from direct searches to HHPred and the BLAST hits from Phagesdb and NCBI BLAST do arise when new information is available through the web services that have not been incorporated into the static PECAAN database for your phage. That is why we have the Last Updated: field under the header for each of these databases. These databases can be Rerun individually for each gene, or under the Admin->Phages menu you can select your phage and press the Edit button for it, which will let you: Reblast all genes (BLAST only) or Rerun Evidence for All Genes (BLAST, HHPred, CDD). Another difference between live and PECAAN data can occur when PECAAN is using an older search database than the online services. You have pointed out some problems previously with the HHPred, which were quite significant. We have therefore tried to increase our database update from monthly to weekly, to try to correspond with their update. The NCBI BLAST and HHPred searches take serious computation and NCBI and the HHPred providers requested that we run these locally on our supercomputer, so that is what we now do, thus the potential for unlinked comparison databases. For HHPred another source of variability is the four databases that we search. We use the CD, Scope70, Pfam-A and PDB databases. There is the option to select other databases in the online execution of this program. The final source of variability in HHPred is built into its architecture. The HMM is built on a set of probabilities and therefore has the possibility of producing slightly different outputs. I have noticed this when two runs produce a hits to the same multi-subunit crystal structure in PDB searches but return different homologous subunits from the crystal. Hope that this helps clarify some of these questions. Thanks, Claire |
Posted in: PECAAN → New Features in PECAAN
| Link to this post | posted 14 Feb, 2019 09:28 | |
|---|---|
|
|
First let me point out that we have added two new fields to the Phagesdb BLAST table, Cluster and Pham. We added these two fields because when considering the functions in Phagesdb BLAST we often found ourselves asking, is this phage from the same sub cluster or is this a gene function that has been acquired from another group of phages? Once we added these fields we found it much easier to sort based on cluster and pham. While clicking on the headers to sort and using the search tool is a wonderful way to explore the Phageseb BLAST information, we found ourselves thinking, wouldn't it be nice if we just had a summary count of the functions along with the cluster and pham information. That is why the summary table was created. The count just shows the number of hits for each function/cluster/pham while the frequency shows the fraction of the total number of phages with an assigned function. When checking genes with no assigned function, NKF, I find it very useful to be able to check the Phagesdb Summary and if there are no function/cluster/pham lines then I know it is not really useful to sort and search the Phagesdb BLAST table. Another interesting feature of this summary table is that it shows the variety of names for the same function. This may be useful to point out to students as a reason for using the "approved function list" represented in the drop down window that appears as functions are typed in. Finally, the Phagesdb Summary is useful to see which clusters may have exchanged this gene and you can use the Pham to make sure that they are fairly close relatives. Hope that this helps and that the new features are useful to you and your students. Thanks, Claire |
Posted in: PECAAN → New Features in PECAAN
| Link to this post | posted 17 Oct, 2018 21:10 | |
|---|---|
|
|
I have a question on what to list as the function for Kalb97 gene 76, which has good matches to the Immunity Repressor genes in other phages. An alignment of Kalb97 with one of it's closest relatives, Marius, show that Kalb97 has suffered a 630 bp deletion at base 44554. This deletion removes about 266 bp from the 5' end of gene 79 of Marius (Kalb97 gene 76), which is the immunity repressor, and about 10 bp from the 3' end of gene 80 in Marius (Kalb97 gene 77). Therefore the termination site is deleted in Kalb97 gene 77, thus allowing the reading frame to continue. The space for a promoter/regulation and the translation start site for Kalb97 gene 76 as well as about half the N-termal end of the immunity repressor coding region is missing and therefore it is doubtful if this phage has any immunity regulation. The Phagesdb entry for Kalb97 lists it as a Temperate phage but the plaque pictures look to be clear plaques. The deletion is also evident in the NCBI BLAST results where the T:Q From is 90:1 for Marius. My question is, what do I list as the function? Do I still put Immunity repressor and then try to put a note in the gene that indicates that this gene carries a clear plaque deletion? Or do I simply label it as NKF and ignore the discovery of the deletion? |
Posted in: Functional Annotation → Truncated Immunity Repressor
| Link to this post | posted 24 Jul, 2018 17:00 | |
|---|---|
|
|
Tammy, We currently do not place the function into the function box in our DNA Master Full Annotation export. Welkin just pointed out to me that the online guide tells how to generate minimal files from the complete notes when functions are recorded in the function field. We will work on adding this feature to the complete file export. Meanwhile, you can use the Export CDS Function button on the export page of PECAAN to export a file that can be copy/pasted into the DNA Master Documentation page which will parse the functions into the notes field. These minimal functions can then be copied en masse into the product or function fields in DNA Master by clicking on the right hand triangle in the Notes field. Thanks, Claire |
Posted in: PECAAN → New Features in PECAAN
| Link to this post | posted 23 May, 2018 17:29 | |
|---|---|
|
|
Ruth Plymale recently asked how to annotate a frameshift in PECAAN. Since this is a new feature in PECAAN, I thought that I would post the answer here also. Since I am currently working on AugsMagnumOpus I will demo how to find and record frameshifts with this phage. First a quick look at the GeneMark coding capacity plot shows the coding capacity for the two parts of the longer frameshifted gene: 15175 – 15573 and 15520-15971. (Fig. 1) Notice that the left half is in the top forward reading frame and overlaps with the right half that is coded for in the bottom forward reading frame. If you go to the Sequence menu in PECAAN and scroll down you will see the six-frame translation. The top three are forward translation and the bottom three are reverse. All potential Start (green) and Stop (red) codons are displayed in the Top and Bottom DNA strands. The amino acid in each translation is oriented over the first base of the triplet codon that codes for it. If you type 15520 into the Seek position: box in the Sequence window and press return, the left side of the window will scroll 15520 (Fig 2). Notice that the green gene highlight is in the bottom forward translation and corresponds to the left gene. Recall that in GeneMark, this was in the top forward frame. So, the correlation in frames between PECAAN Six Frame and GeneMark is as follows: PECAAN Six Frame……….GeneMark Forward Top…………..Forward Bottom Forward Middle………..Forward Middle Forward Bottom………..Forward Top Therefore, we will expect the frame shift to move from the bottom reading frame in the PECAAN Six Frame figure to the top reading frame since in GeneMark the left half was in the top reading frame and the right half in the bottom reading frame. If we look for a potentially slippery spot in the Forward DNA strand we see in the middle of Figure 2 there is a stretch of five Gs that correspond to PGE in the bottom translation and AGGS in the top frame translation. During the frameshift the tRNA with the Glycine under label 1 shifts back one base to correspond with the Glycine under label 2 in Figure 2. Therefore, the Guanine under label 3 in Figure 2 is both the last base used in translation of the bottom reading frame and becomes the first base in continuing translation of the top reading frame. To accurately find the position of the G under label 3 in Figure 2, take and drag the figure to the left until the G, under label 3, is positioned as the first base at the left of the window and then use the number under Start pos as its proper location which is 15549. Using the Mouse pos can sometimes be inaccurate and should be used to get general locations. Therefore the two parts of the frameshifted gene are then defined by the ranges 15175 .. 15549 and 15549 .. 15971. Now, return to the Genes window by clicking on Genes in the top menu. Click on the top Add Gene button and the Add Gene window will pop-up. Enter the Gene Stop as 15971 and ensure that the Forward Orientation is displayed. Click on the Advanced button and another field will appear for you to enter the beginning of the right half of the frameshifted gene. Enter 15549 and press the blue + button. Another field will appear that will allow you to enter the right end of the left half of the frameshifted gene which is also 15549 in this case. Again, click the blue + button and you will see the complete entry shown in Figure 3. At this point click the Add Gene button at the bottom of the window and the gene will be added and you will see join(start..15549,15549..15971) as the gene range for the new frame shifted gene. One last thing. You will need to go to the original gene representing the right half of the frame shifted gene and uncheck the Gene Included box so that you don’t have it included in the genome. You should see it designated as (Not Included) when you look at the gene selection drop-down menu. |
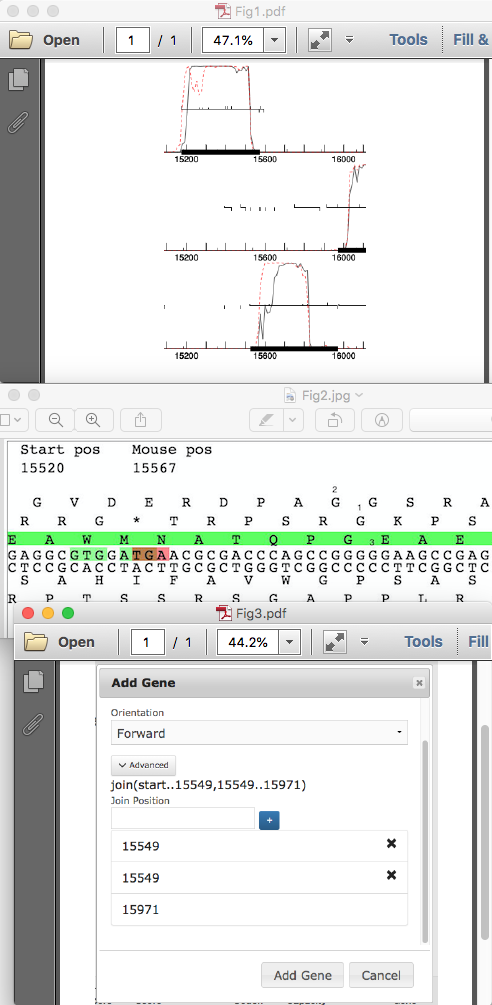 163Kb
163KbPosted in: PECAAN → New Features in PECAAN
| Link to this post | posted 16 Apr, 2018 17:13 | |
|---|---|
|
|
Welkin, So, you are implying that we should not call any gene as a Tail Assembly Chaperone at this point in the B1 Cluster? If we can call genes that match tail assembly chaperones in other B cluster phages, how many can we call, I see some with one, some with two, and some with none? |
Posted in: Cluster B Annotation Tips → Tail assembly chaperones?
| Link to this post | posted 25 Jan, 2018 14:28 | |
|---|---|
|
|
I am working with gene 72 from Niza, which is an A1 Mycobacteriophage. None of the phagesdb genes show a function but the top HHPred match is to pfam 14373, which is an Immunity Superfection protein based on the E.coli T4 gene described in J. Virol 1989 63:3472-8. Abstract below: "The immunity (imm) gene of the Escherichia coli bacteriophage T4 effects exclusion of phage superinfecting cells already infected with T4. A candidate for this gene was placed under the control of the lac regulatory elements in a pUC plasmid. DNA sequencing revealed the presence of an open reading frame encoding a very lipophilic 83-residue (or 73-residue, depending on the unknown site of translation initiation) polypeptide which most likely represents a plasma membrane protein. This gene could be identified as the imm gene because expression from the plasmid caused exclusion of T4 and because interruption of the gene in the phage genome resulted in a phage no longer effecting superinfection immunity. It was found that the fraction of phage which was excluded upon infection of cells possessing the plasmid-encoded Imm protein ejected only about one-half of their DNA. Therefore, the Imm protein inhibited, directly or indirectly, DNA ejection." The pfam14373 description is: "This family includes the E. coli bacteriophage T4 superinfection immunity (imm) protein. When E. coli is sequentially infected with two T-even type bacteriophage the DNA of the superinfecting phage is excluded from the host, into the periplasmic space. The immunity protein plays a role in this process." Looking at Niza-72, the TMHMM predicts three transmembrane domains that covers 71 residues. The total length of the T4 protein is 76 amino acids and Niza-72 is 104 residues. Finally, NCBI has chosen to recently add the pfam14373 region note to all of the top BLAST hits to Niza-72. A decision for inclusion or exclusion of "superinfection immunity protein" in the accepted function list needs to be made since all future annotators of this Cluster A1 protein will want to include this as a function. It looks like Acinetobacter Baumannii (WP_062937363) has already included it as a function. I didn't attach a DNA Master file since it is readily available on PECAAN. Thanks, Claire |
| Link to this post | posted 22 Jan, 2018 18:17 | |
|---|---|
|
|
Sally, I assume that you are referring to Sansa, Aminay, Steamy, Acolyte, and LastResort. I checked on the web Phamerator and could not find them therefore I wonder if they have been Phamerated. PECAAN talks to PITT and St. Louis to pull Phamerator and Starterator information respectively. If a phage hasn't been Phamerated then I don't think that it will show up in the Pitt database. Thanks, Claire |
Posted in: PECAAN → New Features in PECAAN