Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
Debbie Jacobs-Sera posted in Next Phamerator Update?
Steven Caruso posted in Next Phamerator Update?
Debbie Jacobs-Sera posted in Validating Translational Frameshifts in DNA Master
storksle posted in Validating Translational Frameshifts in DNA Master
Debbie Jacobs-Sera posted in Validating Translational Frameshifts in DNA Master
Different coding potential for same sequence?
| Link to this post | posted 14 Mar, 2019 10:37 | |
|---|---|

|
I am curious about how the calculation for coding potential (this is trained to M. foliorum) is determined. We have a called gene that shows no coding potential whatsoever. However, this gene is called in many other phages, and the AA sequence is 100% conserved. In the phages with identical AA sequences, the amount of coding potential varies (from very little to quite a bit). The RBS values don't differ dramatically among the phages. Is this an issue where different runs will give different outputs (I believe coding potential uses a HMM)? Or is GeneMark taking into account non-coding regions adjacent to the gene that may vary? |
| Link to this post | posted 14 Mar, 2019 14:36 | |
|---|---|

|
Evan, Have you read this? https://seaphagesbioinformatics.helpdocsonline.com/article-4 Both Glimmer and GeneMark use a sample of the target genome, so if you run them 10 times you MAY get some differences. Those differences occur most commonly when predicting 'small' genes. It is the primary reason we hand curate the genomes. As for your question, please provide specifics: what gene, what genome, and any other pertinent data. Thanks, debbie |
| Link to this post | posted 14 Mar, 2019 15:24 | |
|---|---|
|
|
Debbie Jacobs-Sera Thanks Debbie. Three examples of the gene are below with screenshots of Stanktossa (no coding potential), TinSulphur (some coding potential) and Superfresh (significant coding potential). All three align 100% (alignments shown are to Stanktossa). All three are on the reverse strand as well. I figured three would be some differences between runs of GeneMark, but since so much weight is put on coding potential, I can see how a gene could be excluded because of those differences. Evan |
 617Kb
617Kb| Link to this post | posted 15 Mar, 2019 01:12 | |
|---|---|
|
|
Evan, Keep in mind that the coding potential is based on a sample of the sequence from each genome, so why would you expect them to be the same? The use of comparative genomics is helpful and necessary here. to prove the point, I just ran GeneMark on Stanktossa and got a different result than you did. Yes, coding potential is very important, but this shows you that it has to be thought of in context. The comparative genomics are impressive. |
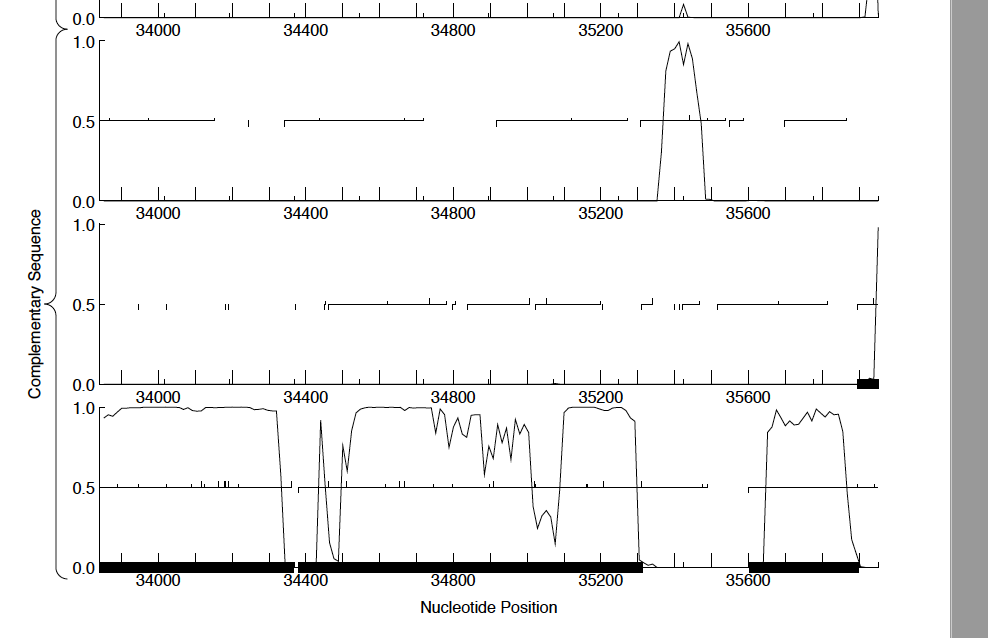 59Kb
59Kb| Link to this post | posted 15 Mar, 2019 01:20 | |
|---|---|
|
|
I guess I'm a little confused then. Isn't the coding potential dependent on the sequence of that part of the genome. All of the regions in my previous post have the exact same amino acid sequence. Even if the sequences are exactly the same, we would expect different coding potentials? |
| Link to this post | posted 15 Mar, 2019 01:22 | |
|---|---|
|
|
No. it is dependent on the random 'sample' that it used. therefore, the patterns picked up work well for big genes, but 'fall apart' for small ones. Yes, the sequence of the gen is the same, but the random samples used to measure against are different. They are even different for the same genome, hence thy are based on a random sample of the genome. |
| Link to this post | posted 15 Mar, 2019 01:25 | |
|---|---|
|
|
Gotcha. So in this situation, since the sequences are highly conserved, we would put more weight on the comparative BLAST results than the coding potential (i.e. - include the gene as opposed to excluding it)? |
| Link to this post | posted 15 Mar, 2019 02:41 | |
|---|---|
|
|
Evan, In this case you can't ignore the comparisons, not can you ignore that there is a gap. If we could say that coding potential is always weighted in a particular way for every gene, we could write the program and stop doing this manually. BUT there are too many factors and they are not weighted the same in each instance (gene), so it requires human evaluation. At least for now…. Do not exclude this gene. (until you have evidence to do so.) |
| Link to this post | posted 15 Mar, 2019 10:06 | |
|---|---|
|
|
Thanks Debbie! |