Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
Gene split in 2
| Link to this post | posted 02 Jun, 2017 21:19 | |
|---|---|

|
In Smairt I have two genes that overlap (ca 550 bp) that both have blastp matches to the same gene porduct in JC27. I have aligned the DNA sequences to that gene and get the following (see attached file as well): Smairt 34B aligns to JC27 gene 37 from base 1 to base 588 with a 11 snps and one indel. Smairt 35 aligns from base 143 to base 688 with 8 snps and one indel. I cannot make Smairt 35 longer to include the first 142 bp with matches in JC27 and 34B cannot be made longer (darn stop codon) to include the last 100 bp that match JC27… None of these putative ORFs have known functions. Call both ORF 34B and 35 in Smairt despite 550bp overlap or just call ORF 34B (which is longer than 35? That is my dimemma |
 2Kb
2Kb| Link to this post | posted 06 Jun, 2017 12:58 | |
|---|---|

|
do both parts have good coding potential? |
| Link to this post | posted 06 Jun, 2017 15:50 | |
|---|---|
|
|
Yes - see attached file - 34B is (obviously) between labeled 34 and 35 |
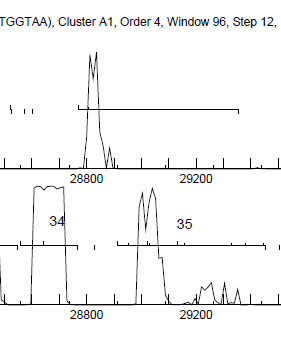 9Kb
9Kb| Link to this post | posted 16 Jun, 2017 19:28 | |
|---|---|

|
This looks very suspicious to me. According to the phage page it was sequenced in 2014 using 454 technology. I would not be surprised if you have a sequencing error here. The 1 indel you are finding is right at a mono-nucleotide run of 6 G:454 technology is notorious for mono-nucleotide run errors (although it usually gets them too short not too long). You should ask Dan to review the assembly in consed (if he has the assembly). I would bet good money the 454 data for that region has some reads with 5 G's and some reads with 6 G's. It is also possible that Consed made an assembly error giving too much validity to low quality reads and mis-aligning all the high quality data, I have seen that happen a few times. I would not be doing any more annotation work on this one until the validity of the sequence in this region is confirmed. |
| Link to this post | posted 16 Jun, 2017 20:30 | |
|---|---|
|
|
Thanks Chris That does look like a possible answer to the problem. I think we may need to go back to BYU for the sequencing data! |
| Link to this post | posted 03 Jul, 2017 15:42 | |
|---|---|

|
Hmmm…I was totally ready to say Chris is right, it's probably a 454 sequencing error, but here is the region in question in the assembly: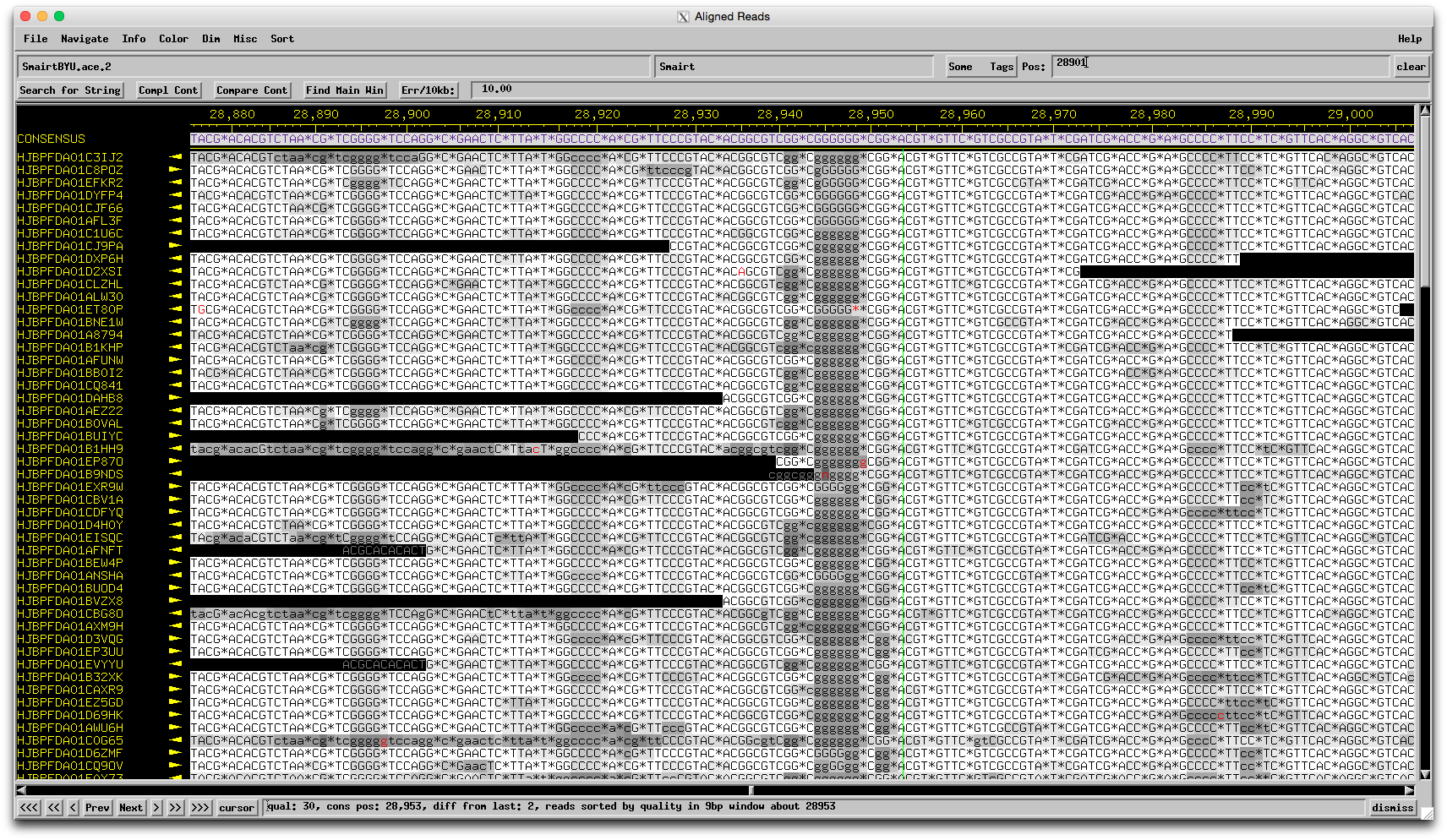 The evidence for 6 Gs there is quite strong, and there are only a few reads that suggest only 5 Gs. That doesn't mean it's not truly 5 Gs, but I wouldn't feel comfortable changing the sequence on this evidence alone. I guess that means you should proceed as is? What do you think Welkin/Chris? –Dan |
| Link to this post | posted 03 Jul, 2017 16:55 | |
|---|---|
|
|
I would go with the sequence as is then (i.e. 6 G's). The sequencing error hypothesis was always a bit of a long shot since the consensus was too long and as I mentioned 454 typically makes consensus errors by making mononucleotide runs too short. As for annotation, that is up to Veronique & Welkin. |
| Link to this post | posted 03 Jul, 2017 17:30 | |
|---|---|
|
|
I would still want to call both genes - the "original" gene in other phages as no known function. The question is whether I annotate both as "hypothetical protein" or "hypothetical protein, N-terminus" and "hypothetical protein, C-terminus". And what Genbank will accept… |
| Link to this post | posted 07 Jul, 2017 19:17 | |
|---|---|
|
|
Dan, is it worth doing a Sanger run across the area to resolve? |