Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by uOttawaPHAGE
| Link to this post | posted 18 Apr, 2025 02:55 | |
|---|---|
|
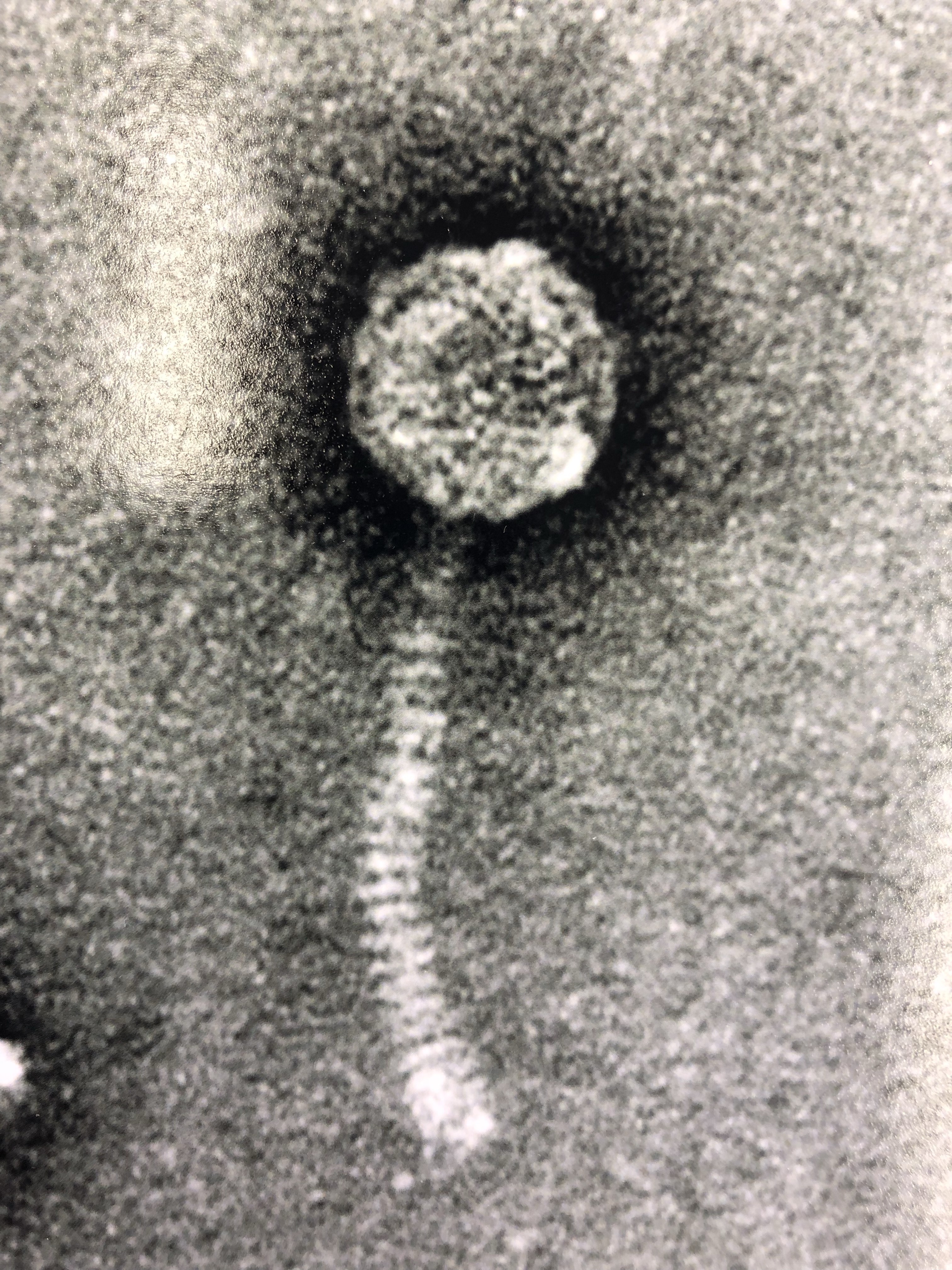
We would like to propose the function "Fin anti-sigmaF factor" should be added to the Functional Assignment list. Zippen_60 (43340 bp- 43552 bp) may be a "Fin anti-sigmaF factor" Sequence is: MAQREVITFICDNCGGEDDLKEGQILPTGWMEILFTENSQANEPVQTCQLCTDAVAKALAERRGDIAPSK Currently 47 members of this Pham from a variety of clusters. Some annotated as "DNA binding protein" Support for the function: 1. Two HHPred hits, greater than 96% probability and high coverage, to "Anti-sigma-F-factor" (see attached image or go to the HHPred Job https://toolkit.tuebingen.mpg.de/jobs/1090560 Hit 1: PF10955.11 - http://pfam-legacy.xfam.org/family/PF10955.11 Hit 2: PDB 5MSL - https://www.rcsb.org/structure/5MSL 2. Protein has two CXXC motifs spaced about 35 resides apart. These conserved cysteine pairs are critical for coordinating a Zn²⁺ ion in anti-sigma factor domains which bind Zinc and is similar to the zinc-binding anti-sigma factors Fin and CsfB from Bacillus subtilis (see the paper doi:10.1111/mmi.13724) 3. MSA alignment for the pham shows that two conserved CXXC motifs (see attached image) 4. Alphafold3 predictions of Fin and Zippen_60 are somewhat similar, and predicts Zn binding by the four cysteines in Zippen_60 and strong probability for dimerization, similar to Fin. See Attached images. |
 1.32Mb
1.32Mb| Link to this post | posted 28 Feb, 2025 05:10 | |
|---|---|
|
|
hi Debbie OK, it sounds like it is OK for one minor tail protein to be significantly further downstream from the others. We recognized it had an activity found in tail proteins and hits with other tail proteins. We will call it as a minor tail protein. Well aware that the TEM misled the student into thinking this was a podo. We quickly saw that it was a myo and will likely repeat the TEM before the end of the term. thanks Adam |
| Link to this post | posted 25 Feb, 2025 22:38 | |
|---|---|
|
|
We are annotating phage Circuit and aren't sure how to call Circuit_Draft gene 31 (26021 - 27280). This Pham is called minor tail protein in several clusters including in comparator FH phage Bumble. However, in some phages it is called Hydrolase (for example Vibaki_31, cluster FL), and other FH phages have a different Pham in same position often called as a hydrolase (Klevey_34). Circuit has three minor tail proteins after the tape measure, and then ~10 genes before gene 31, so we weren't sure if this arrangement is OK, or if all the minor tail proteins need to be adjacent to each other. |
| Link to this post | posted 08 Jun, 2024 13:58 | |
|---|---|
|
|
Kieran Furlong, a student in my group, created this Observable notebook to help harmonize ~30 AZ genomes. It can be used for any cluster. Give it a try! https://observablehq.com/d/0fd237126fd99985 Adam |
| Link to this post | posted 08 Feb, 2024 03:25 | |
|---|---|
|
|
Hi Debbie The question and the evidence has been generated by a student in my course. I think the second gene (25,700 to 26,641) should be annotated as an exonuclease. Clear coding potential, strong functional hits, annotated in all comparators, most used start site that covers all coding potential. I'm not sure what to do for the first gene. Either: 1) Annotate the gene (25,195 to 26,052) as a membrane protein, respect the coding potential and allow a 353 overlap. Two other phages annotate this gene, but as I mentioned above the encoded genes are shorter. No hits on Blastp or HHpred to mention. 2) Leave a gap before the exonuclease and call nothing. I'm skeptical the auto-annotated bottom strand calls are real: only very weak GenemarkS coding potential (dotted red lines)and no matches on HHpred or Blastp. The coding potential is much higher on the top strand. 3) I suppose a third option would be a translational frameshift, though we'd obviously need experimental evidence to make that call. I didn't see any evidence for a slippery site near the transition in coding potential between the frames. I think the decision hinges on whether a 353bp overlap would be acceptable. It severely violates a Guiding Principle. I haven't annotated enough genomes to have a sense what the correct decision is. thanks! Adam |
Posted in: Annotation → Gap or overlap in Superstar (BD2)
| Link to this post | posted 07 Feb, 2024 22:08 | |
|---|---|
|
|
Hi Debbie Continuing this thread with a different, but related question about Superstar. The genes in question are between 25,200 and 27,000. The auto-annotation added two bottom strand genes, but we see stronger coding potential on the top strand, and we think we should call the gene (a membrane protein with no significant HHpred hits) from 25,195 to 26,052. However, this gene has a very large overlap with the next gene (an exonuclease). To preserve coding potential, use the best RBS scores and match the Glimmer/Genemark calls, the best call for the second gene is 25,700 to 26,641. This would create a 353 bp overlap between the two genes. A related phage, Caelum in the attached document, also has a gene in the same pham on the top strand (gp31 in the attached phamerator map), but with a smaller overlap (~50bp). Would a 353bp overlap be acceptable? Or does the large overlap of the gene at 25,195 to 26,052 in Superstar suggest that the similar gene in Caelum (and Issmi) might have been incorrectly called, and all three should be removed? This would leave a gap and unassigned coding potential. We can move the start site of the second gene, but the start at 25,700 is the most frequently called start and creates a gene in Superstar of similar size to those in related phages. thanks! Adam |
Posted in: Annotation → Gap or overlap in Superstar (BD2)
| Link to this post | posted 07 Feb, 2024 21:17 | |
|---|---|
|
|
Hi Debbie Starting this thread up again with a question from a student, Ashlyn: For draft phage Superstar (BD2), the gene at the coordinates 31,549 to 31,764 (gene 47 on Phamerator) is currently being called a DNA binding protein. However, HHPRED shows high coverage and probability for a helix-turn-helix DNA binding protein. We would like to be as specific as possible for this call. According to the SEA-PHAGES FUNCTIONAL ASSIGNMENTS sheet, the alignments of the protein hitting HTHs must hit 2-3 alpha helices in the sequence separated by small spacer (turn) regions of 3-4 amino acids. However, the alignments I found are showing a spacer region of 6 amino acids between the 2nd and third helix. Is it acceptable to call this as a HTH, or should we simply call it a DNA binding protein? Attached are the top HHpred hits and the sequence comparison for the top hit. thanks Adam |
Posted in: Annotation → helix-turn-helix binding domain or protein?
| Link to this post | posted 07 Feb, 2024 13:04 | |
|---|---|
|
|
Hi Debbie Ok, this was a misunderstanding on my part. I thought we needed to identify a slippery sequence similar to those that have been defined previously (i.e. on the table). CCCTTTTT definitely works as an XXXYYYZ sequence, and is in the correct position for a -1 frameshift. PF to PF referred to the slip as coding proline-phenylalanine to proline-phenylalanine. thanks Adam |
Posted in: Cluster AY Annotation Tips → Tail Assembly Chaperone
| Link to this post | posted 07 Feb, 2024 07:04 | |
|---|---|
|
|
hi We are annotating phage Aikyam. We noted that phages in this cluster with the same TAC pham, phage Isolde, for example, call the programmed frameshift using a slippery sequence CCCTTTTT (a PF to PF -1 slip). CCCTTTTT doesn't appear on any list of validated slippery sequences that I could find, so I wasn't sure why this site has been called. It certainly works as a slippery sequence, but is there any evidence to support this call? Other AY phages, like EvePickles, use CCCAAAAA (PK to PK; a different Pham), which does appear on Sup Table 1 from Xu et al. thanks Adam |
Posted in: Cluster AY Annotation Tips → Tail Assembly Chaperone
| Link to this post | posted 02 Feb, 2024 17:30 | |
|---|---|
|
|
Hi Debbie The student is carefully annotating this region, and I understand the functional information could influence the start site call, but what if these were two NKFs? I'm interested in the more generic case because it would change the way I think about the annotation guiding principles regarding overlap. If they were NKFs, would I make the same call and use the 44271 start, accepting the 158bp overlap? Based on the coding potential, I think I would want to. Even though Issmi has dissimilar nucleotide sequence, the GeneMark files looks quite similar for all three phages: good coding potential but a lot of overlap. I probably would have included the gene and I was wondering if there is a reason it wasn't changed in QC that I'm missing. Based on synteny, I suspect we'll find the same pham in Issmi (though we haven't checked). A |
Posted in: Annotation → Gap or overlap in Superstar (BD2)