Welcome to the forums at seaphages.org. Please feel free to ask any questions related to the SEA-PHAGES program. Any logged-in user may post new topics and reply to existing topics. If you'd like to see a new forum created, please contact us using our form or email us at info@seaphages.org.
Recent Activity
All posts created by MSMC
| Link to this post | posted 12 Jun, 2023 14:56 | |
|---|---|

|
Pollenz Thanks Rick, I was wondering about the C-terminal domains when I was looking at this. I agree, I'll stick with the HTH domain call. |
| Link to this post | posted 05 Jun, 2023 17:55 | |
|---|---|
|
|
Pollenz Rick, do you still believe these should be called as immunity repressor? I'm QCing an FF phage now, and I've got two genes back to back that align well with the immunity repressor and excisionase. There are two tyrosine integrases present in the genome. I'm inclined to call this immunity repressor and excisionase (taking into account your argument above), but I don't know if there has been a change in consensus since this thread (https://seaphages.org/forums/topic/4785/). |
| Link to this post | posted 19 Aug, 2022 14:53 | |
|---|---|
|
|
I'm having the same problem. I just upgraded my MacBook to an M1 chip. I'm running Windows 11 on Parallels. I've downloaded DNA Master, the folder is on my desktop (the name is “DNA Master”. When I run the program as an administrator, it gives me an error message "Invalid File Name", which then disappears. When I try to load an existing archive file, or even just start DNA Master without a file, the "TbCodonBias: Cannot perform this operation on a closed dataset" error appears (so I can’t open the program at all). It also give the following error (most times, not every time): “Queries table is corrupt. Reload from FTP DNA Master FTP site” I've tried uninstalling and reinstalling to a different directory, but it still defaults to the desktop. Any help would be great! |
Posted in: DNA Master → TbQueries
| Link to this post | posted 19 Jul, 2022 18:18 | |
|---|---|
|
|
We are using the Wizard DNA Clean-Up System kit for DNA extraction (as we have for many years), and recently our DNA samples have had extremely high 260/280 ratios (most between 2.6 to 3.0). Things we've tried: 1. New Kit (Guanididium and Columns) 2. New Water Eluent 3. New EDTA 4. New SDS 5. Syringe vs. Vacuum manifold Our spec seems fine, as other samples give normal ratios. The A260 seems high (vs. low 280), as when we run samples on the gel, the mass appears lower on the gel than calculated by the spec. We're running out of ideas, any thoughts? |
| Link to this post | posted 13 Jan, 2022 22:07 | |
|---|---|
|
|
jawsWPI We do something very similar with peer review, using a Google Doc for students to record their annotation data. |
Posted in: Notes and Final Files → Notes format when using DNA Master
| Link to this post | posted 21 Jul, 2021 18:16 | |
|---|---|
|
|
cdshaffer Thanks Chris! I've been going gene by gene in PhagesDB, which has wworked, but I'll try the admin page strategy as well. |
Posted in: PECAAN → PECAAN not showing Starterator and Pham info
| Link to this post | posted 19 Jul, 2021 10:38 | |
|---|---|
|
|
Is there a reason that PECAAN provides a link to the Starterator report and Pham page for some genomes, but not others? See screenshots below - Looper is a Gordonia phage, Eraser is an Arthro phage. Cheers, Evan |
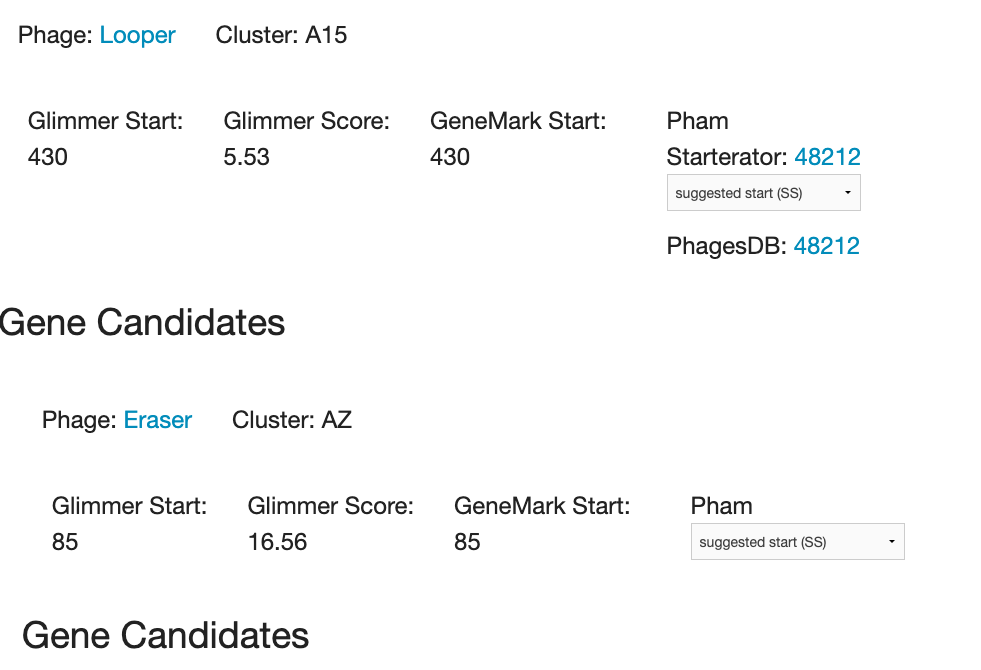 75Kb
75KbPosted in: PECAAN → PECAAN not showing Starterator and Pham info
| Link to this post | posted 15 Jul, 2021 14:32 | |
|---|---|
|
|
DanRussell Great, thanks so much for checking this Dan! |
Posted in: Annotation → Is this a sequencing error?
| Link to this post | posted 14 Jul, 2021 19:40 | |
|---|---|
|
|
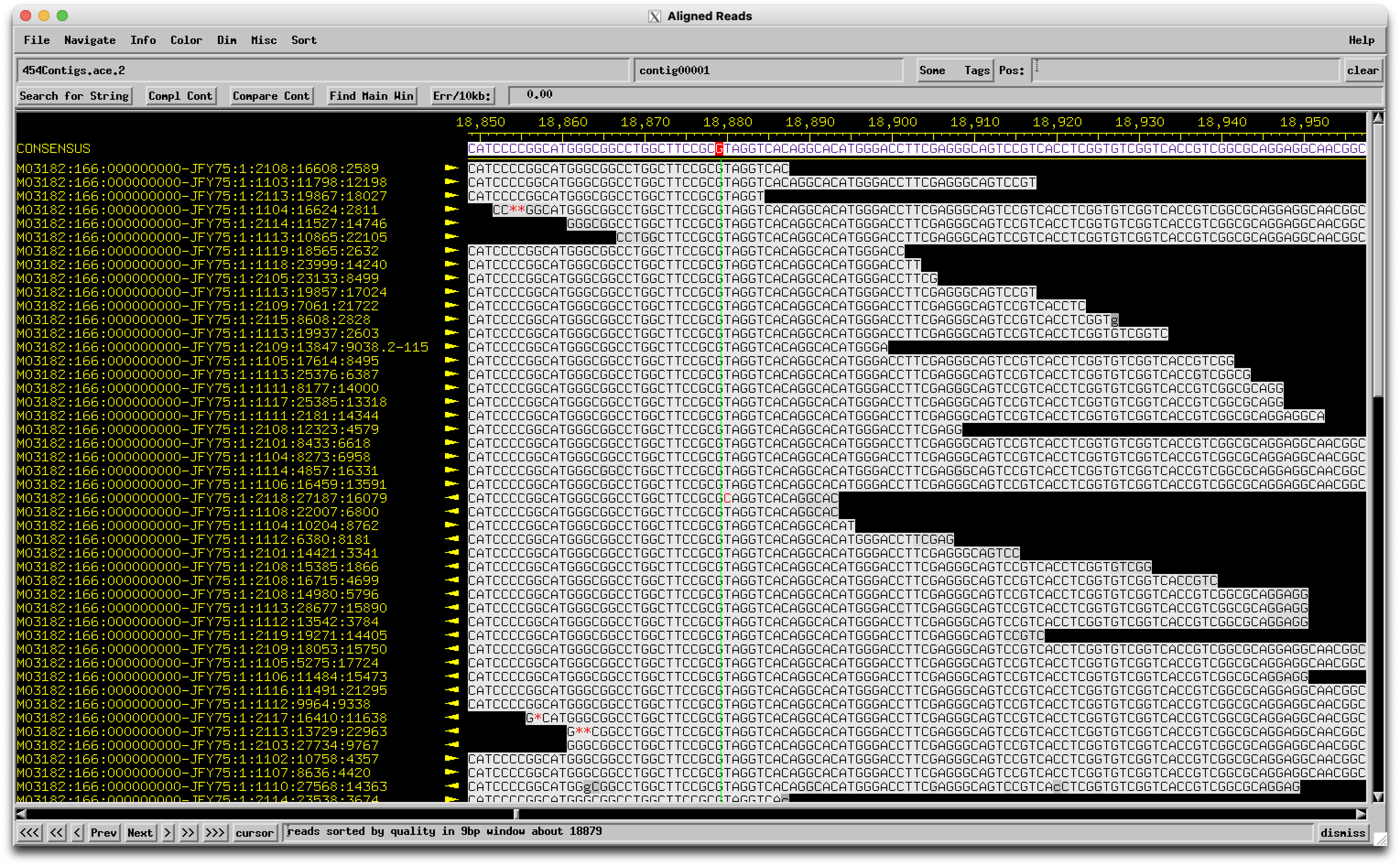
We are annotating genes 8 and 9 in Looper (A15). These two genes are currently orphams. When we look at the same region in similar phages, it's highly conserved and called as one gene (Pham 69334 - 723 members of the pham). When the region from Looper is BLASTed, the alignment shows one nucleotide difference compared to similar phages (see attached), and this results in a stop codon - therefore 2 genes in Looper and one gene in the other phages. There is little coding potential for the second gene in Looper (both programs call it a gene), but the coding potential is minimal for the second half of the gene in similar phages. So the question is, is this a sequencing error? It seems unlikely that one phage out of hundreds would have a mutation leading to a stop codon resulting in two functional genes. |
 78Kb
78KbPosted in: Annotation → Is this a sequencing error?
| Link to this post | posted 18 Jun, 2020 15:07 | |
|---|---|
|
|
jawsWPI Thanks Joanne, I was thinking the same thing, I kept the third gene, there are some hits moderate hits to collagen-like proteins. The annotators did include the HNH. |
Posted in: Cluster A Annotation Tips → minor tail proteins